



May 5, 2026

What are New Approach Methodologies for Drug Safety Assessment?
NAMs are more human-relevant, non-animal studies that fall into four main categories:
- Microphysiological systems (MPS): 2D/3D cultures and organ- or organoid-on-chip platforms that mimic human tissue function.
- Advanced in vitro assays: Tools such as cytokine-release and T-cell activation panels to assess immunotoxicity.
- Advanced ex vivo human systems: Including tissue culture and pluripotent stem cells for high-throughput safety screening.
- In-silico tools: Computer-based models that simulate drug absorption, distribution, metabolism, excretion (ADME), off-target effects, and immunogenicity.
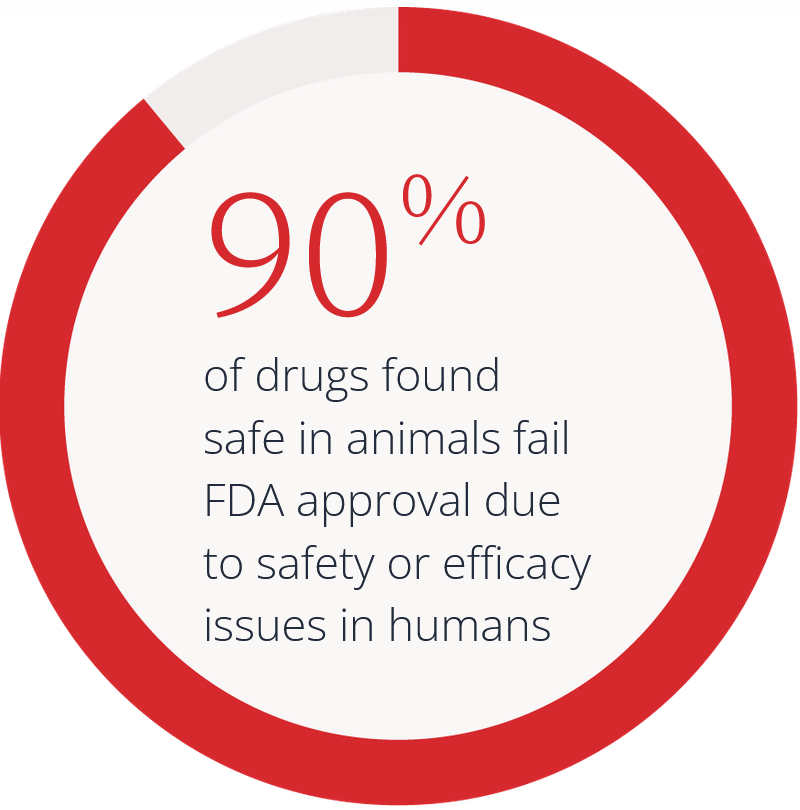
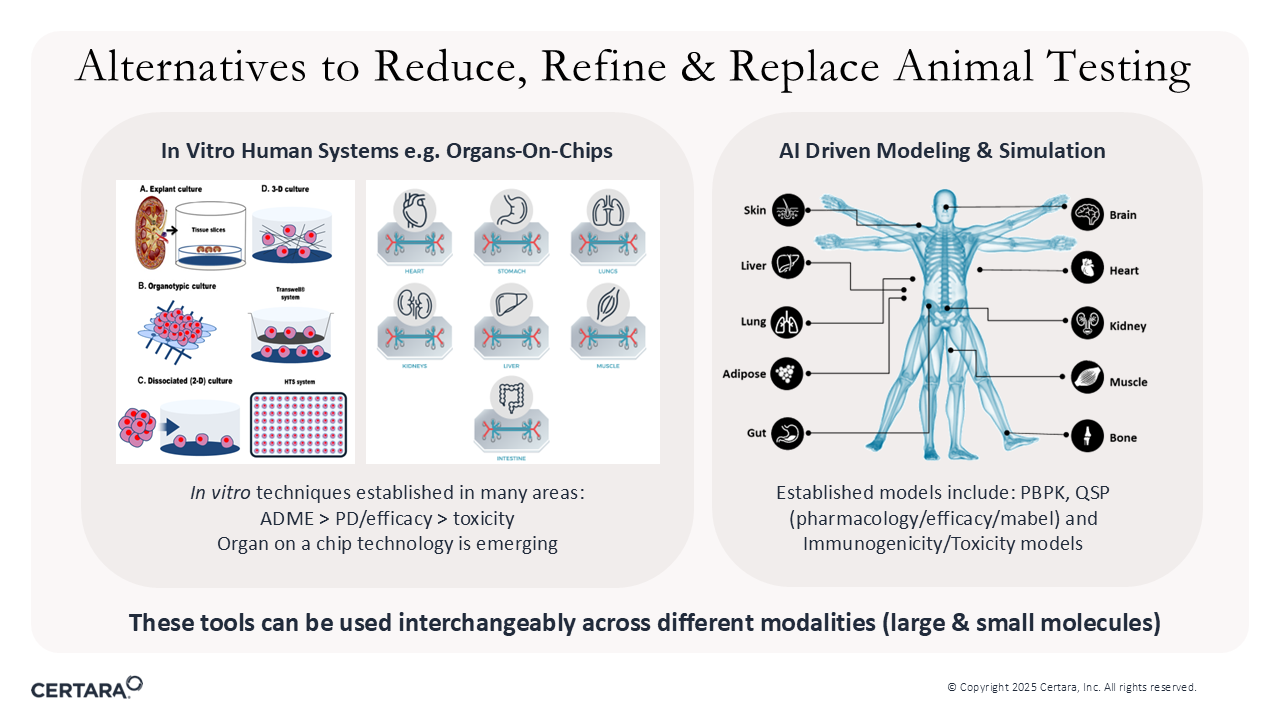
Figure 1. NAMs are alternatives to reduce, refine, and replace animal testing.



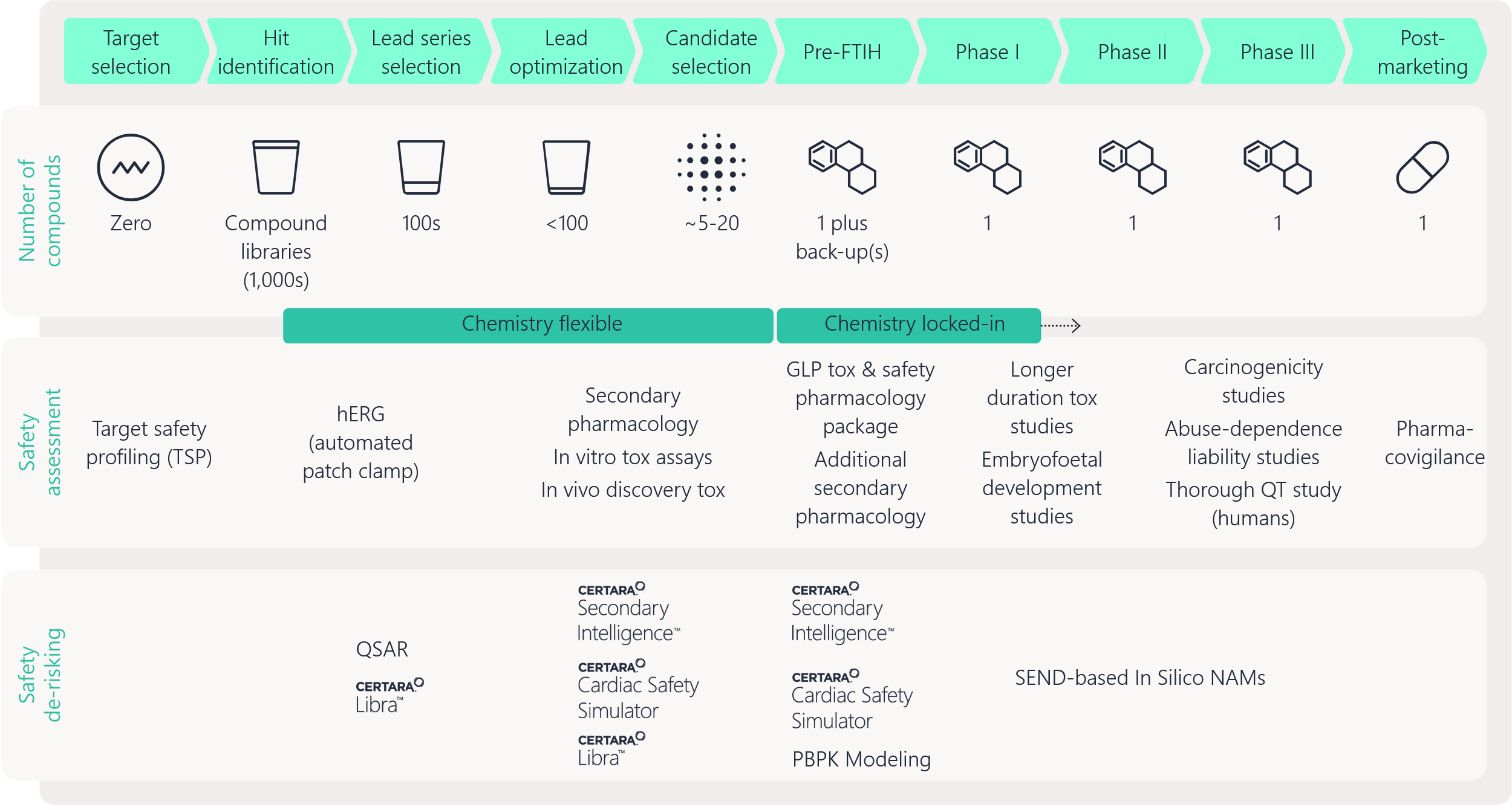
Figure 2.Opportunities for in silico NAMs throughout drug development


Figure 3. Drug developers must use a weight-of-evidence approach to develop their regulatory submissions for health authorities.
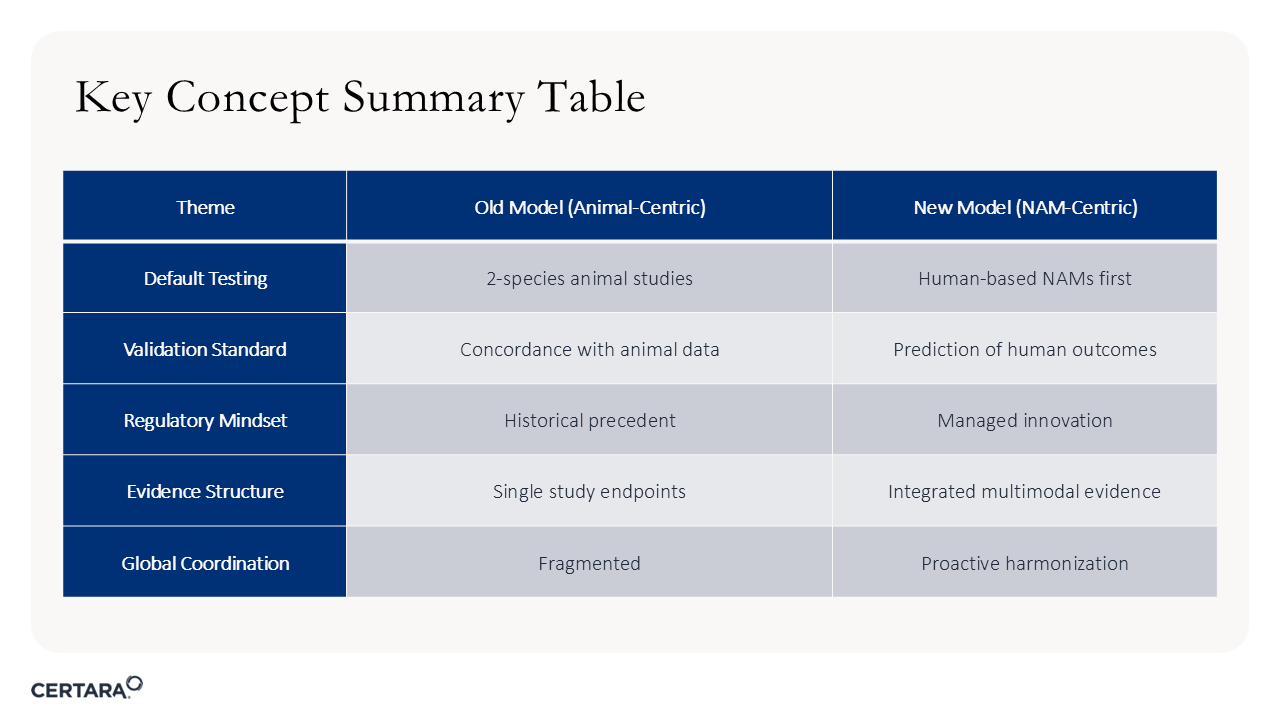
Figure 4. Shifting from animal-centric drug development to a NAM-centric model requires multiple considerations.
Monoclonal antibodies are leading the way in eliminating animal testing
The FDA’s eventual goal is to eliminate animal testing for all drugs. But the FDA Roadmap prioritizes reducing non-human primate testing for monoclonal antibodies (mAbs), potentially eliminating an average of 144 NHPs per drug at $50K each. The nonclinical testing of mAbs presents unique challenges due to their precise target specificity, which often means there are no suitable non-human species for study, or NHPs may be the sole viable option.

To facilitate regulators accepting NAMs in mAb programs, companies can focus on a few key steps. These recommendations are particularly relevant for “me too” mAbs that target the same proteins as existing approved therapies. If the target and antibody are already well understood, more animal testing may not add much value.
By carefully comparing the new mAb to approved ones, examining its structure, binding mechanism, potency, and behavior in the body, and utilizing in vitro data and other robust evidence, companies can demonstrate that the safety risks are already well understood. This makes it possible to reduce or skip some animal studies, especially for long-term and reproductive toxicity. The goal is to only use animal studies when truly needed. Otherwise, sponsors should leverage existing clinical data as well as lab and in silico model results to fill knowledge gaps.
- Age
- Weight
- Tissue Volumes
- Tissue Composition
- Cardiac Output
- Tissue Blood Flows
- [Plasma Protein]
- Dose
- Administration route
- Frequency
- Co-administered drugs
- Populations
- No of male/female
- MW
- LogP
- pKa
- Protein binding
- BP ratio
- In vitro Metabolism
- Permeability
- Solubility
Figure 5. Schematic for physiologically-based pharmacokinetic modeling
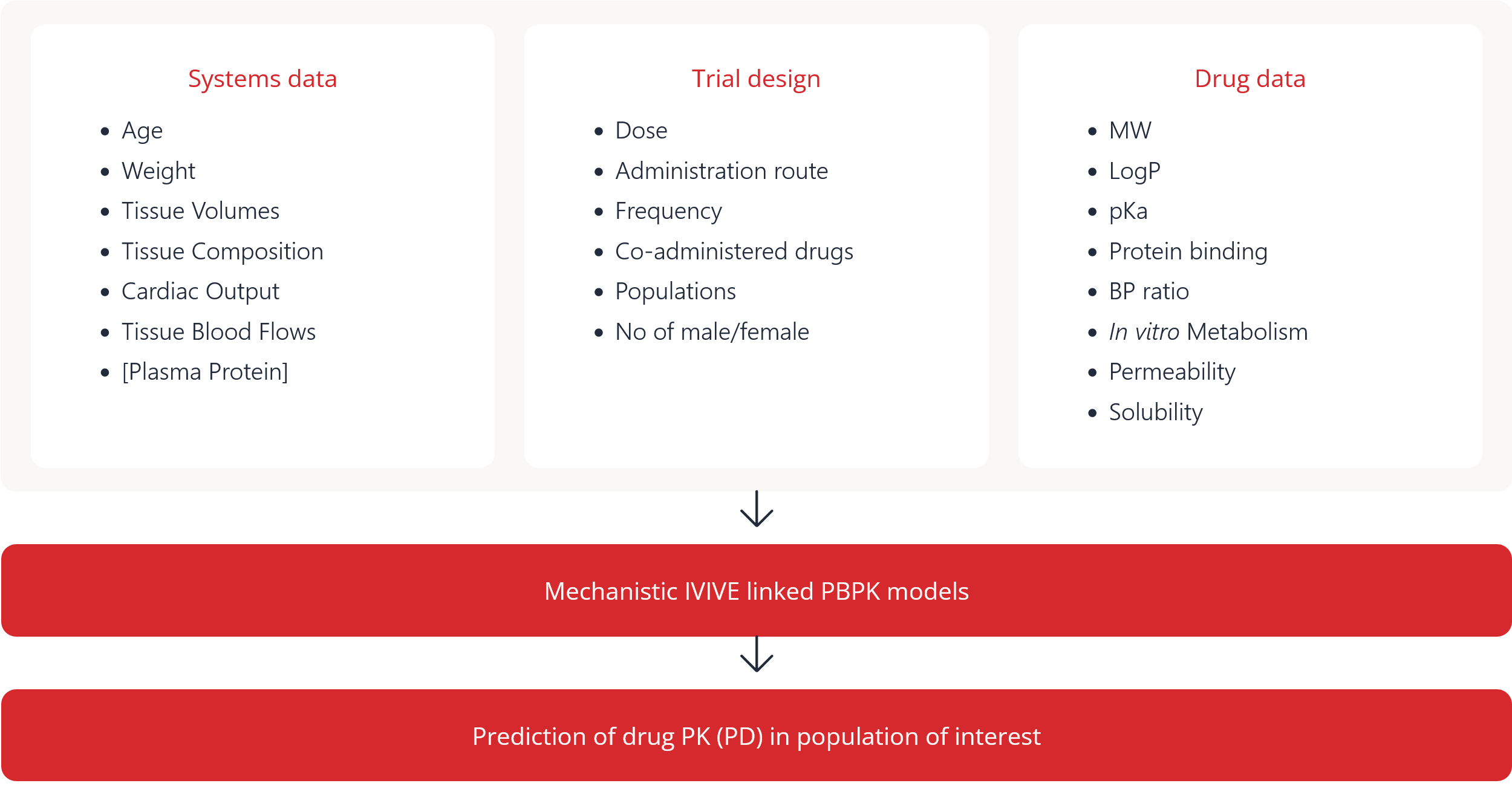

Figure 6. What type of New Approach Methodology have you used in preclinical drug development programs? N=200

Are you prepared for the FDA’s phase out of animal tests for mAbs?
The FDA’s plan to phase out animal testing is a transformative step that paves the way for innovative, human-relevant preclinical approaches that are more predictive, efficient, and ethical. There is a long history at the FDA of using validated new approach methodologies (NAMs), including in silico tools and computational modeling as a framework for regulatory decision-making to support this transition effectively.

Suzanne Minton
Director of Content StrategyDr. Suzanne Minton is the Director of Content Strategy where she leads a team of writers that develop the whip smart, educational, and persuasive content is the foundation of Certara’s thought leadership programs. She has a decade of experience in corporate marketing and has conducted biomedical research in infectious disease, cancer, pharmacology, and neurobiology. Suzanne earned a BS in biology from Duke University and a doctorate in pharmacology from the University of North Carolina at Chapel Hill.
Author’s note: this blog post was originally published in July 2025 and has been updated for accuracy and comprehensiveness.
End of Animal Testing? Advancing Drug Development Alternatives
The STAT article, written by Prof. Amin Rostami-Hodjegan, explores the transition from animal testing to alternative drug development methods. It highlights the limitations of traditional animal models, such as their inability to reliably predict human outcomes, and the ethical and logistical challenges they pose.

Contact our expert consultants


