


July 13, 2026

Want to learn more about designing HI studies and applying PBPK modeling? View the full webinar.
Learn more
Certara’s clinical pharmacology and PBPK experts support sponsors across all stages of drug development, including hepatic and renal impairment study design, modeling and simulation, regulatory strategy, and label negotiation.


Zoe Barter, PhD
Senior Director, PBPK Consultancy, CertaraDr. Barter has over 20 years of experience in drug metabolism, transport, complex interactions, and specific populations with a strong background in in vitro to in vivo extrapolation (IVIVE). She received her PhD in Drug Metabolism from the University of Sheffield in 2005. This was followed by a joint position as a Postdoctoral Researcher within the Unit of Clinical Pharmacology at the University of Sheffield and Research Scientist at Certara. In 2009, she joined Certara full-time where she has been involved in projects related to extrapolating clearance and extending compound and population databases within Simcyp Simulator and modeling of complex transporter and metabolism mediated drug-drug interactions and inter-ethnic differences in pharmacokinetics.

Nolan Wood, PhD
VP, Clinical Pharmacology ConsultingDr Nolan Wood joined Certara in 2017 as a consultant in the clinical pharmacology and translational medicine group, providing clinical pharmacology leadership and stewardship to develop high quality, innovative clinical pharmacology programs supporting global drug development. Dr. Wood has more than 35 years of global drug development experience working in the pharmaceutical industry and as a consultant, covering all stages of development, from first-in-human studies through to regulatory submissions, including pediatric drug development. He has worked across a wide range of therapeutic areas involving both small molecules and biological products. Working as the global clinical pharmacology lead, he has taken responsibility for implementing model-informed drug development strategies resulting in successful global regulatory approvals for several compounds. He has contributed to the design and execution of studies in special populations including patients with renal and hepatic impairment and pediatric populations. Dr. Wood has extensive experience of interactions with the major health authorities and has written Clinical Pharmacology sections for regulatory submissions and requests for information.
Following a degree in Pharmacology, Dr Wood obtained a PhD in the Clinical Pharmacology Group at the University of Southampton. He has (co-) authored over 40 peer-reviewed publications and is a member of the British Pharmacological Society.
FAQs
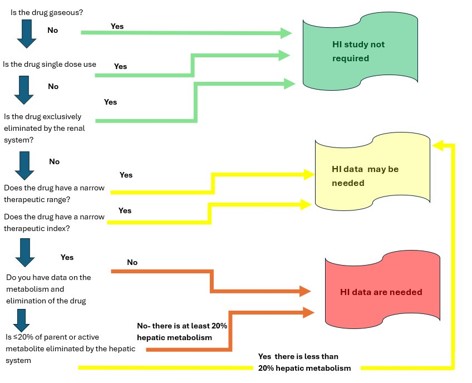
When is a hepatic impairment study not required?
A dedicated hepatic impairment study is generally not required if the drug is gaseous or volatile (eliminated via the lungs), if hepatic metabolism or biliary excretion accounts for less than 20% of the absorbed dose, or if the drug has a wide therapeutic index with limited hepatic involvement. However, each case should be evaluated against the applicable regulatory guidance, and teams should be prepared for the possibility of a post-marketing commitment if a study is not conducted.
What is the difference between a full design and a reduced design hepatic impairment study?
A full design study enrolls patients with mild, moderate, and severe hepatic impairment alongside matched healthy controls, providing comprehensive data across the entire spectrum of dysfunction. A reduced design study includes only moderate hepatic impairment and controls. The reduced design is a common first step, but if a meaningful PK change is observed, sponsors may need to extend to severe or mild groups, depending on the regulatory agency.
Can PBPK modeling replace a dedicated hepatic impairment clinical study?
In some cases, yes. Both the FDA and EMA have accepted PBPK modeling in lieu of, or to supplement, clinical HI studies, particularly for extrapolating from single to multiple dose, from one Child-Pugh category to another, or to derive dose recommendations when clinical data is limited. However, acceptance is more likely when the drug’s ADME properties are well-characterized, clinical PK data is robust, and the predicted exposure shift is clinically manageable. Severe hepatic impairment is often held to a higher bar and may still require a post-marketing study.
What is the Child-Pugh scale and why is it used in HI studies?
The Child-Pugh scale is a widely accepted clinical scoring system used to assess the degree of hepatic impairment. It incorporates three laboratory measures and two clinical measures to classify patients as Child-Pugh A (mild), B (moderate), or C (severe) impairment. Most regulatory guidance for HI studies references Child-Pugh classification for enrollment criteria and for defining the degree of impairment being studied. In oncology settings, the NCI Organ Dysfunction Working Group (NCIODAG) classification based on bilirubin and ALT may also be used.
How early in development should hepatic impairment be considered?
Hepatic impairment planning should begin during nonclinical development, where early in vitro and in vivo ADME data can indicate the extent of hepatic metabolism. If significant hepatic elimination is anticipated, eligibility criteria for early phase 1 studies can be designed to include patients with mild HI. Building in pre-specified PopPK sampling from the outset maximizes the value of that data and may reduce the need for a dedicated standalone study later.


