



Cancer treatment has evolved from generally cytotoxic therapies towards drugs that target specific pathways and alterations in cancer cells. Patients tolerate targeted therapies better, and their treatment continues over a period of years, instead of a few weeks. These improvements in cancer treatment spurred the FDA’s Project Optimus initiative which has altered the criteria used to determine the appropriate dose of a drug through development away from this historic maximum tolerated dose (MTD) paradigm.
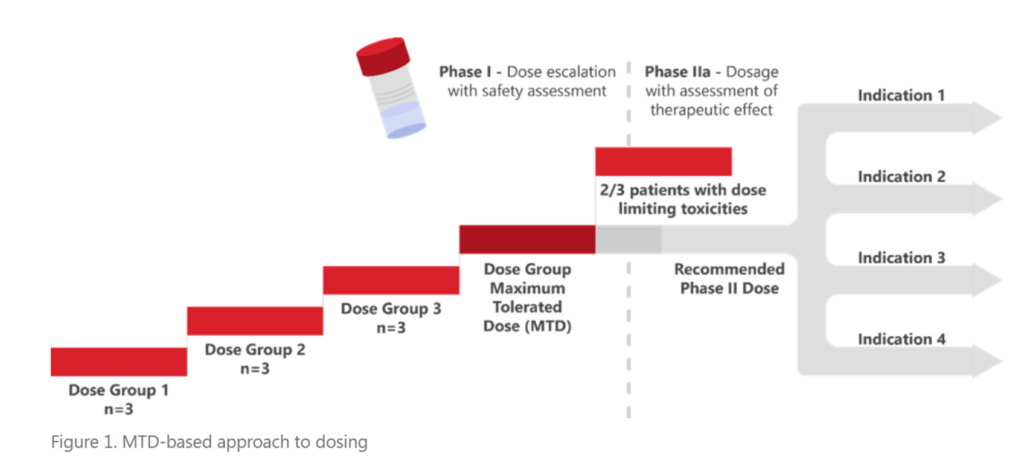
In May 2022, the American Society of Clinical Oncology (ASCO) and FDA jointly presented an initial workshop discussing the challenges and strategies around dose optimization of agents used as monotherapy (read about it in this blog, Highlights from the FDA-ASCO Workshop on Dose Optimization for Oncology Drugs). This second joint ASCO/FDA workshop (conducted on September 6/7, 2023) discussed concerns, issues, and ideas for the optimization of doses when multiple drugs were being administered.
Below are summaries of the key take-home messages from this meeting:
1. Balancing toxicity is key: There was strong consensus that for combination regimens, a consideration of the tolerability of the overall regimen, and not just its efficacy, is essential. This is especially relevant since efficacy gains in oncology populations might be limited, but overlapping toxicities are often significant. In other words, while the combination might not be synergistic from an efficacy perspective, toxicities might indeed be, rendering the overall risk-benefit of the combination unfavorable. Practically this means that the appropriateness of both the dosage of the investigational agent and the combination agent may need to be further assessed and justified in the combination setting. A key implicit but obvious takeaway is that the dosage of both agents needs to be considered, even when the combination partner is a previously approved agent.
Given this need, a key question posed for discussion was “How much monotherapy data is needed before optimization of combination dosage can begin?” Beginning evaluation of the combination therapy early in development can decrease the total number of patients needed and help to determine the appropriate safe and efficacious doses more quickly. However, the FDA stated that sponsors should wait until the toxicities and tolerability associated with the monotherapy of a new drug are identified before adding additional drugs to the regimen. There was an acknowledgment that less monotherapy evaluation may be needed in situations where the investigational agent will never be used outside of a combination setting (e.g., drugs for which monotherapy activity is expected to be limited or if there is no development path for the agent as a monotherapy). Decisions regarding the amount of monotherapy data required need to be made on a case-by-case basis and appropriately justified.
2. Dose optimization burden of proof ‘grids’: Multiple speakers presented decision trees to aid with determining the extent of dose optimization that will be needed for combination development (Figure 2). All considered the approval status of the combination agent and the mechanism of the combination as either synergistic or additive. In cases where there is no overlapping toxicity, and the drug activities are separate, it may be possible to ‘fix’ the approved drug in combination and optimize only the investigational drug (new molecule entity; NME). However, if the toxicities are overlapping, or if the mechanisms are likely to interact, it may be necessary to modify the dosages of both the approved drug and the investigational drug to optimize the dose regimen. To help streamline the process, speakers recommended that sponsors prioritize which agent to optimize the dose for first (e.g., the more potent agent that contributes primarily/more to efficacy can be optimized first as was done in the dabrafenib case study presented).
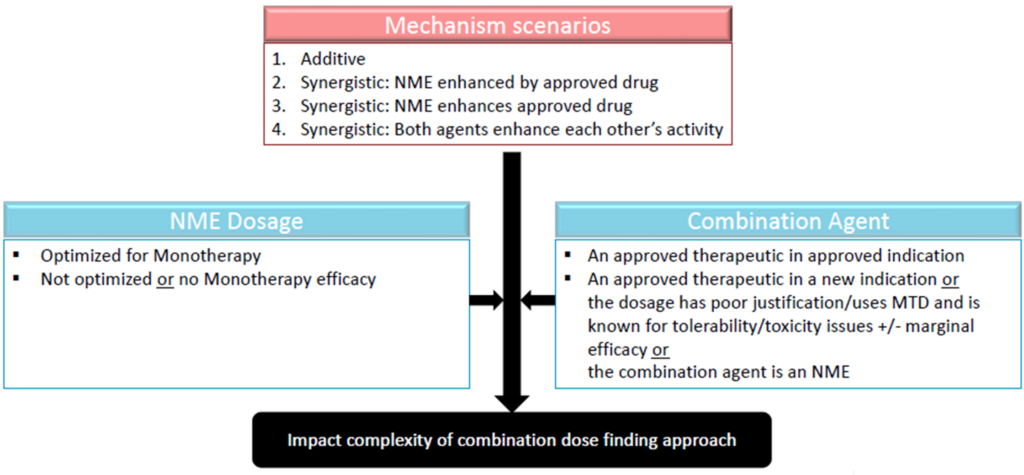
Figure 2. Decision trees on dose optimization needed for oncology drug combination development. Source: Adapted from the presentation by Dr. Julie Bullock
3. Leveraging non-clinical data and translational modeling better: Consistent with the practice in non-oncology fields, the concept of exploring activity and toxicity vs dose or exposure ‘surfaces’ was discussed during the workshop (Figure 3). These ‘surfaces’ can be more extensively explored, and the optimal clinical dose or exposure range of both agents can be narrowed in the non-clinical space to guide dosage optimization of the combination therapy.
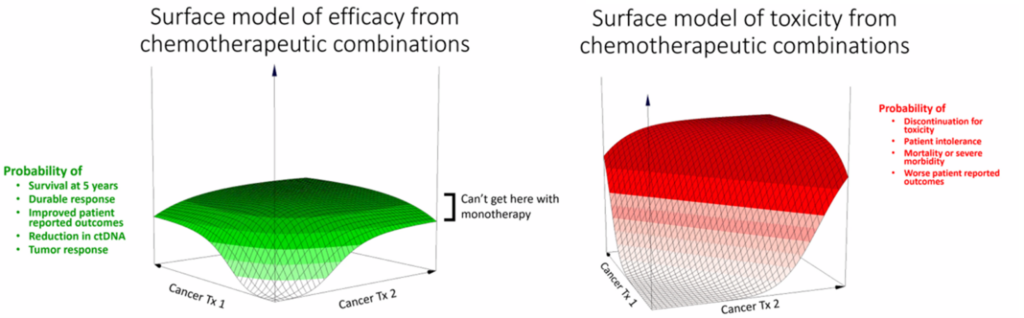
Figure 3. Surface models for drug efficacy and toxicity for chemotherapy combinations. Source: Adapted from the presentation by Dr. Steve Schafer
Currently, non-clinical testing of drug combinations is typically limited to pharmacology and growth inhibition models. However, these models often lack dose-response and rarely evaluate the impact of dosage or timing of dosing. There are opportunities to improve how we interrogate combinations in non-clinical settings to enlighten how we approach clinical dose optimization strategy and dose selection. In addition to better dose-ranging data, the need to evaluate not just inhibition of tumor growth, but also the duration of the response was recommended. This could be accomplished by extending the duration of the observation period in non-clinical studies. In addition, a case example was provided wherein testing clinically relevant, feasible, or representative regimens in non-clinical models could rule out certain combination partners, further enlightening the combination clinical development approach.
4. Understanding exposure-response (E-R) is central to decision-making on combination dosing: The common thread in all three case examples presented was that the sponsor embarked on combination dose optimization studies with a hypothesis regarding benefit-risk based on known exposure-response relationships of the monotherapy agents. In addition, the combination dosages chosen were all determined based on E-R analyses, further validating the importance of this tool. Dr Atik Rahman, Division Director of the Division of Cancer Pharmacology-1 (DCP‑I ) at the FDA Office of Clinical Pharmacology (OCP) also stated that applying model-informed drug development (MIDD) to combination dose optimization is currently underutilized and can be improved. Combining E-R models into ‘surface models’ as highlighted above is one improvement that should be considered.
5. More complex study designs are inevitable for combination dose optimization moving forward: Many novel designs that can aid with determining the MTD and optimal dose range of combination agents, including a Bayesian Optimal Interval (BOIN1) design for combination trials, BOIN-COMB, BOIN-ETC, and a Multi-arm, Multi-stage for combinations (MAMS) design were presented. Opportunities for lead-in studies that seamlessly test monotherapy and combination therapy (e.g., with monotherapy as a lead-in to combination therapy) were also discussed. Intra-patient adaptive dose designs that allow escalations/de-escalations within patients can be explored. Combination dose optimization strategies will need to be determined on a case-by-case basis; while the acceptability of any of these strategies remains to be determined, all may be considered with appropriate supporting data. Nevertheless, because of the expected increasing complexity of combination development programs, early engagement, and multidisciplinary collaboration between clinical, clinical pharmacology, pharmacometrics, and statistics will become increasingly important.
To learn more about how Certara’s clinical pharmacology experts can help you navigate the FDA’s Project Optimus, visit this webpage.
1BOIN designs are statistical methodologies for phase I dose finding clinical trials where the goal is to find the maximum tolerated dose (MTD) or the optimal biological dose (OBD) of an investigational drug based on safety and efficacy.
