



The FDA-ASCO (American Society of Clinical Oncology) workshop on oncology dose optimization (3-5 May 2022) provided the most in-depth view to date into the FDA’s thinking regarding the Oncology Center of Excellence (OCE) led Project Optimus and the shifting paradigm of dose optimization in oncology. Below are a few key take-aways from this meeting:
Project Optimus is most definitely a ‘go’
The seriousness of the FDA’s intent was perhaps best expressed by Dr. Rick Pazdur in his opening remarks: “Nobody has the right or franchise to expose patients to unnecessary toxicities.” Dr. Pazdur clearly stated that past FDA decisions around acceptability or adequacy of early (primarily maximum tolerated dose or MTD-based; Figure 1) dose evaluation are not indicative of future decision-making regarding dose optimization for oncology programs.
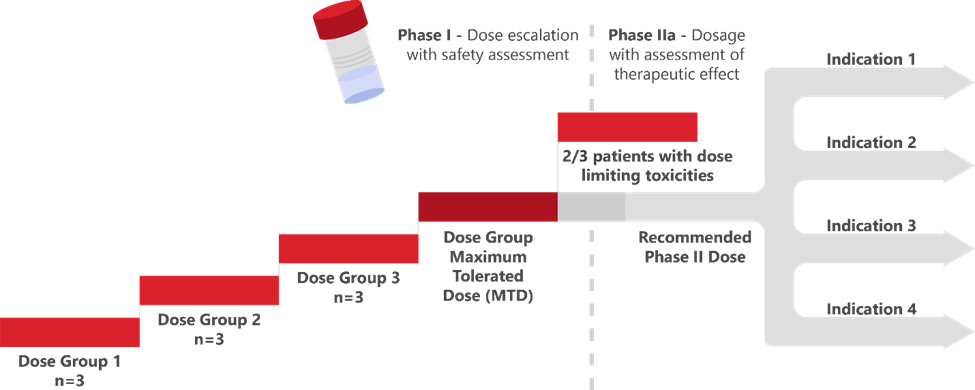
Some have raised concerns that an increased investment of resources in early dose optimization could slow the progress of key therapies to market. In response, the Agency, supported by patient advocates, stressed the importance of ensuring that patients can make informed decisions regarding their dose and are able to stay on these lifesaving or potentially curative drugs once they are on the market without significantly compromising their quality of life. The Agency pointed to the change in the development environment for HIV drugs in the 1990s as an example of a therapeutic area successfully acting on a similar shift in expectations around dose optimization.
Based on discussions at this workshop and consistent with prior messaging from the Agency, Project Optimus-related dose finding due diligence will be expected for all small molecule and therapeutic biologic assets, but the extent of the diligence will depend on the individual asset’s complexities. The Agency indicated that the initiative will permit right-sized development programs that ensure adequate data are available early to enable termination of unpromising drugs while allowing promising candidates to progress to registrational trials with a regimen most likely to achieve a good risk-benefit balance and ultimately result in a successful development program.
A paradigm-shift and added flexibility in early oncology program design
Consistent with this outlook, multiple sessions on the first day highlighted updated frameworks for early oncology drug development program design and pointed to a shift away from historical phase 1/2 nomenclature in oncology towards a more objective-focused nomenclature for studies. One of the key objectives of early oncology trials moving forward will be a more careful understanding of dose- or exposure-response relationships of a drug with both safety and anti-tumor activity – in a relevant patient population, prior to initiation of potentially registrational trials.
At the heart of this early dose optimization remains an evaluation – ideally randomized – of at least two dosing regimens. These regimens are selected after thorough vetting of the totality of clinical pharmacokinetic (PK), pharmacodynamic (PD), safety, anti-tumor activity data from early dose escalation and cohort expansion, supported by non-clinical evidence. Workshop presenters discussed multiple approaches to enable efficient progress through early dose finding studies, including accelerated titration, algorithm-based dose escalation study designs, use of backfill cohorts, improved use of non-clinical data, and model-informed drug development (MIDD) to inform dose selection and use of innovative approaches to improve early trial enrollment, including increasing historically underrepresented patients’ access to trials.
A patient-centric approach to review of drug toxicities
Consistent with previously communicated intent, the FDA focused on retiring dose decision-making based purely on DLTs during the DLT monitoring period. They also reinforced that later onset and lower grade toxicities, particularly those that are symptomatic and impact patient quality of life, should not be ignored. This could imply a shift away from trivializing adverse events (AE) such as diarrhea and fatigue and might indicate that toxicities, such as GI toxicities that were often historically ignored as part of dose decision-making, might receive renewed review attention.
The discussion on patient reported outcomes (PROs) during the second day of the workshop underscored the importance and utility of PRO data as a more frequent, and perhaps more representative, means of capturing a patient’s experience of drug toxicities. Patient advocates at the workshop expressed the importance of fully understanding the consequences of dose reduction, so that patients can make an informed decision whether to tolerate a high burden of toxicities for the promise of better efficacy. Interpreted another way, this may mean the ‘toxicities were well managed with dose modifications’ argument is unlikely to be acceptable moving forward unless there is compelling evidence that to receive the benefit of a particular therapy, those toxicities are unavoidable.
Multiple oncology drug trial design questions remain
Questions remain post-workshop regarding key topics such as determining the appropriate sample size for randomized dose evaluation cohorts, dose optimization approaches for therapies with limited monotherapy activity, the need for dose optimization for an investigational drug in each new indication, burden of dose optimization proof for new drug combinations, etc. These questions were not resolved during the workshop, possibly to allow flexibility on the part of both sponsors and the Agency that permits data-driven decisions on a case-by-case basis for each program.
Nonetheless, a key take-away was that, ultimately, sponsors need to fully understand the dose- and/or exposure-response curve for both safety and efficacy for each drug in the context of its clinical use. Some of these questions will require clinical data to be fully addressed, while some may be answered by leveraging scientific literature or translational information. The agency confirmed its willingness to engage with sponsors early in development and to partner flexibly with them to help address these issues as development proceeds.
To learn more about how Certara’s experts can help you navigate the FDA’s Project Optimus, visit this webpage.
Project Optimus