



The safety of human participants in clinical trials, the intended patient population, and for the labeled indications at the point of introduction into the market is undoubtedly the paramount consideration of any drug hunting enterprise. Often considered the distant (and unfortunately often forgotten) cousin of its more popular version, “efficacy”, safety can be relegated to the back seat. In other words, while sponsors push to understand dose/response for efficacy, sponsors rarely worry about safety if there is general or “acceptable” safety (Figure 1).
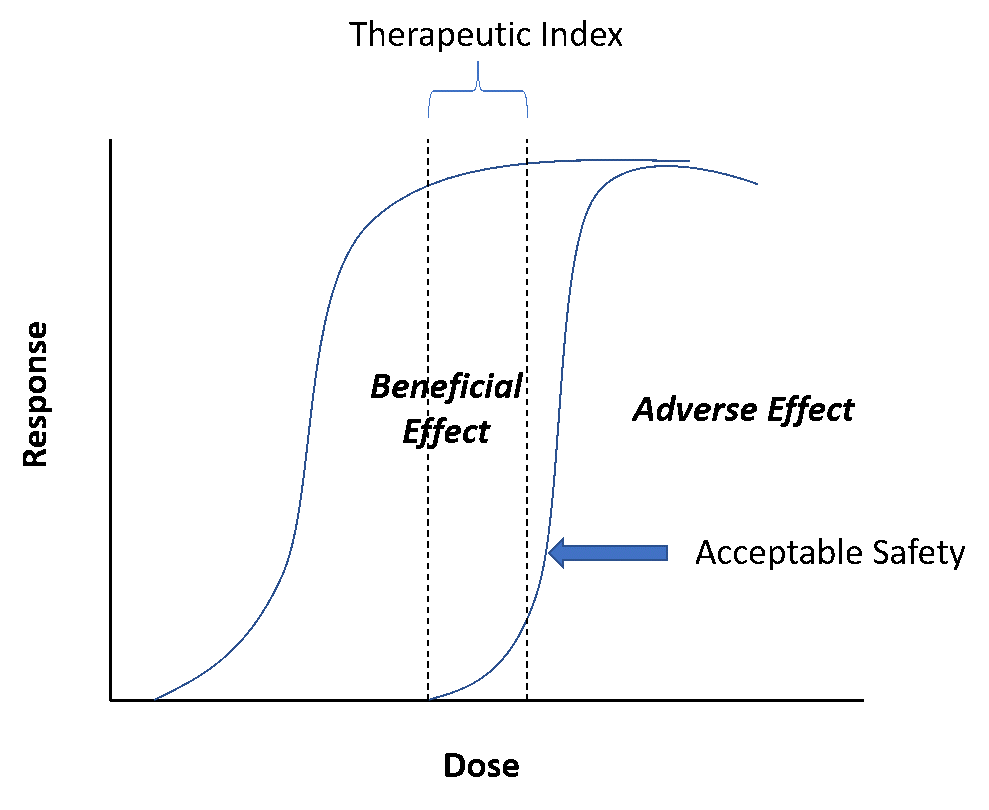
Figure 1: Illustration of the concept of acceptable safety
What does acceptable mean? Well, that’s a nebulous term. In general, if there are no “issues,” safety is often not the center of attraction, not even worth reflecting about. This is where, unfortunately, most mistakes are made. If anything, safety should be given far more attention than efficacy.
Small signals seen in early development, say from time of entry into humans to achieving proof of concept, can be warning signs that can emerge and even derail the program in late development. Pay careful attention to this aspect! Even more important is to keep efficacy within the context of the safety dynamic range. This boils down to the therapeutic window and the all-important question – “Is there a therapeutic window?” Or, in a more proximal regulatory sense, “Is there acceptable risk/benefit?” In a “dose high first” culture, subtle safety aspects can sometimes be exaggerated, and understanding the pharmacology behind it can prepare you and your team for a good handle on this distant cousin.
This blog will reflect on three things drug developers should pay attention to with regards to safety, especially when taking a new chemical entity or biologic drug into human testing.
Watch for emerging preclinical warning signs
In drug discovery, as you look for druggable targets, you will conduct some due diligence on the type and nature of the target you are pursuing. Very likely, there may be some precedent around the target. If you compare the drug pipelines of large pharmaceutical companies, you might notice a homology of about 30% in terms of the targets companies tend to pursue. One would have a general idea to expect some off-target activity. This will be tested extensively in a battery of preclinical pharmacology studies. From those tests, preliminary signs of what to expect in terms of warning signs, be they mechanism-based or compound-based, will start to emerge.
As you enter the preclinical toxicology development program, several safety assessment studies are both needed and typically done which offers further opportunities to learn about key safety signals. These studies are often categorized by duration of drug exposure and include, for a small molecule, single dose (acute) studies, multiple dose studies (subacute, typically 2-4 weeks; and sub-chronic, typically 3 months), and chronic (6-9 months). Both rodent and non-rodent studies need to be performed. These studies are instrumental in identifying any potential dose- and/or exposure-related safety signals in various tissues, organs, and organ systems. When identified, the studies also assess whether the toxicity is reversible. For more details, refer to the ICH M3 document entitled “Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals.” This is just one guidance but there is a plethora of documents relating to nonclinical studies. Such general toxicology studies are complimented by safety pharmacology batteries of tests for various organ systems.
Conventional wisdom dictates that these generalized toxicity studies be performed in at least two animal species, and guidances specify that one be in a rodent and the other be in a non-rodent species. This is to ensure that one develops a comprehensive risk assessment that is without regard to species and as such depends on the wisdom that by testing it in two phylogenetically different animal species one can discern a potential safety finding. When developing biologics, often a single species is sufficient. This is because species selection depends on specificity of epitopes. As such, a second species may be rendered irrelevant. Species selection is critical and routinely rationalized based on pharmacology, selectivity, and pharmacokinetics (Morton, 1998).
Now, these studies with defined parameters and rules of engagement produce a large depth of data which often sits idle when no meaningful signs emerge. When this happens, one tends to question whether the cost-benefit is meaningful. A new field of predictive toxicology (FDA 2020, Wang et al., 2021) has emerged in recent years which specifically questions the validity of our assumptions and whether there are more targeted inquisition of safety issues during drug development. The intent is to deconstruct complex information streams into meaningful biological and toxicological information. As reflected by pundits in this field, the scope of predictive toxicology depends on the first principles of systems biology, and by doing that relying on some novel tools and techniques that use bioinformatics and machine learning to provide an integrated assessment (Wang et al., 2021).
Use in-silico tools and model-informed drug development tools to predict potential showstoppers
The emergence of complex streams of data in the preclinical phase use model-informed drug discovery tools to help make sense of data. For example, the types of questions such tools often provide scenarios and options for, include questions like – what is the probability the molecule I am developing has a chance of increasing blood pressure in a clinically meaningful way? This is a hard question to answer in a nonclinical study, especially for a molecule that is yet to be tested in the clinic. For example, it took years for Pfizer’s torcetrapib molecule to show clinically meaningful increases in blood pressure to the extent that it could lead to adverse safety issues in humans. Refer to the Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events (ILLUMINATE) trial, which was a large Phase 3 trial which was terminated in December 2006 due to all-cause mortality in the torcetrapib treatment group. Obviously, the goal is to prevent the progression of molecules into human testing with such harmful effects much earlier than what happened for torcetrapib (Funder, 2010). In this regard, Certara’s own Secondary Intelligence™ software assembles, curates, and visualizes all secondary pharmacology analyses, providing key information on potential adverse events (AEs), quantitatively rating each compound as to its likelihood of causing off-target safety issues that could impact clinical progress (Figure 2).
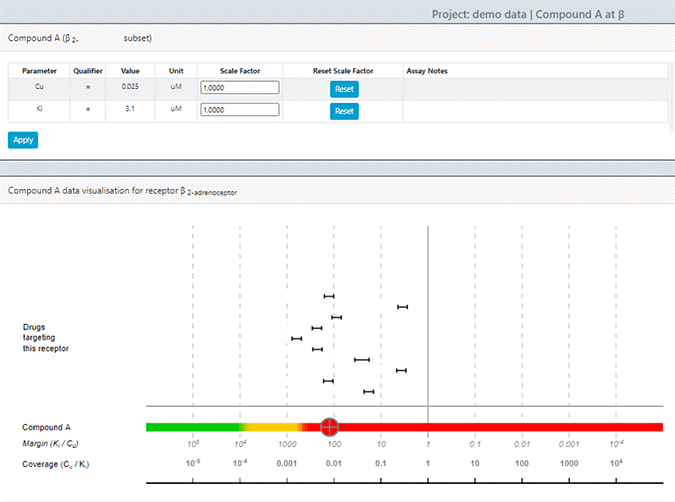
Figure 2: Screenshot from Secondary Intelligence
Carefully monitor safety from the first-in-human trials onwards
Once in first-in-human (FIH) trials, you can further establish guidelines on assessing and monitoring the safety of clinical trial participants. Obviously, multiple stakeholders will provide inputs on your protocol, including the health authority, the clinical site where the study is conducted, as well as the IRB/ethics committees. If you are contending with unique safety issues, you may develop stopping rules for both subjects and panel treatments (whether dose adjustments downwards or termination). You might also elect to include data safety monitoring boards or committees to make independent assessments. The advantage of assessing safety in FIH trials includes the fact that you are likely exploring a wide dynamic range, and perhaps you might identify a maximum tolerated dose. Because you will collect pharmacokinetic data, you can relate the safety observations with drug exposure and develop hypothesis generating assessments. These will help you define your MIDD plans.
There are principally two types of FIH trial designs, one that allows within-panel ascending dose design, and the other is a cohort-based ascending dose design. In the former, the same subjects are washed out before exposing them to a higher dose. In the latter, an entire new subject cohort is exposed to the next higher dose. The former design affords less information loss, in that it gives you a within-subject data reads with each escalating dose. One can assess trends in vital signs or laboratory safety as dose increments progress. One can decide if those trends look alarming enough to scale back a higher dose or to repeat a dose to see if those trends are meaningful. Keep in mind that you have a placebo group that will allow you to see cohort-based blinded review of the trends in safety tests.
In addition, drug-induced cardiovascular adverse events are one of the leading causes of drug withdrawals from the market and of drug label restrictions. Ensuring time-matched collection of 12 lead Holter (ECGs) with pharmacokinetics will set you up well for concentration/QT interval analysis (see Figure 3) to assess the risk of drug-induced cardiotoxicity. And that will assist in assessing whether you have a meaningful QT prolongation signal to worry about. If you don’t see a signal of concern, you could argue against doing a dedicated thorough QT study. If you do see a signal of concern, you can incorporate sufficient ECG safety monitoring in your later phase clinical studies.
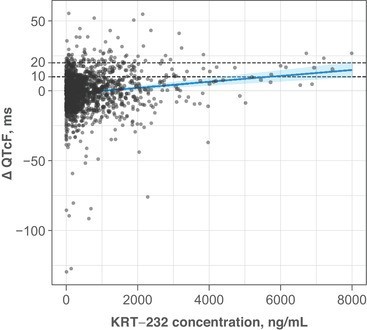
Figure 3: Example of a concentration-QT relationship (Adapted from Taylor et al., 2021)
Depending on worrying trends you might have seen in the FIH study, you may design appropriate safety studies for early clinical development. Taking the torcetrapib example, if you have a follow-on molecule that you think is a differentiator from torcetrapib, you could study your molecule in a 24-hour ambulatory blood pressure monitoring study to see if you have meaningful signals. Using pharmacokinetic/pharmacodynamic (PK/PD) analyses, you can then assess the probability of observing a “torcetrapib-like” effect with your molecule. That study can inform the next decision regarding the developability of your molecule.
In summary, a plethora of emerging tools can quantify the probability of seeing an adverse effect based on the preponderance of data collected during nonclinical and early clinical development. One such tool is Secondary Intelligence, which reduces uncertainty in off target activities that may stall your development program. To learn more about this tool, please watch this webinar.
References
Dorato MA, McMillian CL, Vodicnik MJ. The toxicological assessment of pharmaceutical and biotechnology products. In: Wallace Hayes A, editor. Principles and methods of toxicology. 5th ed. CRC Press; 2008. p. 325–67.
FDA 2020. FDA’s Predictive Toxicology Roadmap. URL: https://www.fda.gov/science-research/about-science-research-fda/fdas-predictive-toxicology-roadmap. Last accessed: December 30, 2021.
Funder JW. Aldosterone, Sodium, and Hypertension Lessons From Torcetrapib? Hypertension. 2010;55:221-223.
Morton DM. Importance of species selection in drug toxicity testing. Toxicol Lett 1998;102–103:545–50.
M3(R2) nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. International Conference on Harmonization (ICH).
Taylor A et al. Phase 1 Concentration‐QTc and Cardiac Safety Analysis of the MDM2 Antagonist KRT‐232 in Patients With Advanced Solid Tumors, Multiple Myeloma, or Acute Myeloid Leukemia. Clin Pharmacol Drug Dev. 2021 Aug; 10(8): 918–926. Wang MW et al. Machine Learning in Predictive Toxicology: Recent Applications and Future Directions for Classification Models Chem. Res. Toxicol. 2021, 34, 2, 217–239.
