


As a pharmacologist, I’ve always been curious about how drugs work. A favorite time-killer while waiting at a doctor’s office is reading drug labels (also known as package inserts) contained within the pages of “lifestyle” magazines.
Ever wonder what a package insert looks like? Ask a pharmacist.
By law, pharmacies are supposed to offer that to you, but hardly any commercial pharmacy ever parts with this without pressing. I am not sure why. You can try to navigate your local health authority website, and if you’re lucky, you may find the elusive package insert. Alternatively, some drug manufacturers’ websites will carry these elusive documents, but you may find a more shortened variant. If you are lucky to find one, look at the “Mechanism of Action” section. That should tell you how your drug works. In some cases, this section may say “The mechanism by which this drug works is unknown.” One wonders why somebody would want to take a drug for something if you don’t know how it is supposed to work.
In this blog, I will reflect on 3 key questions a drug developer must consider when developing a new molecular entity (NME).
Do you understand the biology?
This is probably the first thing you must ask about your NME. Sometimes, understanding the biology is straightforward, but often, it’s not.
For example, consider diabetes. If you’re developing a next generation dipeptidyl peptidase-IV (DPP4) inhibitor, then you may be in luck. There is a plethora of DPP4 inhibitors so it may be straightforward to understand the biology. Quite simply, a selective DPP4 inhibitor targets the incretin axis in the treatment of patients with T2D. You might have run studies in rodents that are bred to be genetically deficient in DPP4 to see if there is improved glucose tolerance, elevated active GLP-1 levels, increased insulin secretion, and decreased circulating glucagon. There may be further data showing that your molecule does not have anything to do with DPP-8/9. Your challenge may be to come up with sustained DPP4 inhibition and consistent increases in active GLP-1 and GIP levels, to make sure your gliptin is better than those currently in use.
Alternatively, you may be an immunologist working on developing the next generation checkpoint inhibitor as a novel oncology drug. You may have a drug that is designed to block checkpoint proteins, which work as gatekeepers to immune responses. Blocking these checkpoints (such as CTLA4, PD1, or PDL1) allows the T cells to attack the tumor cells. Now, this biology is more complex as it deals with the whole system of tumor microenvironment, and the complex interactions between cells, proteins, and immune components.
Do you understand how your molecule is designed to work?
A drug can work at its site of action in several ways. Most often, it engages with a receptor directly (orthosteric site), forms a drug-receptor complex, and then elicits the desired or sometimes undesired effects. Pharmacology exists to explain how drugs engage with their presumptive targets. These drug-receptor interactions are key to understanding the kinetics of drug action (Figure 1).
Agonist drugs bind to the receptor and activate it. On the other hand, antagonist drugs bind to the receptor and prevent other drugs from binding and eliciting a desired response. Inverse agonists bind to and prevent receptor activity when the agonist is not present. A partial agonist drug cannot fully activate the receptor.
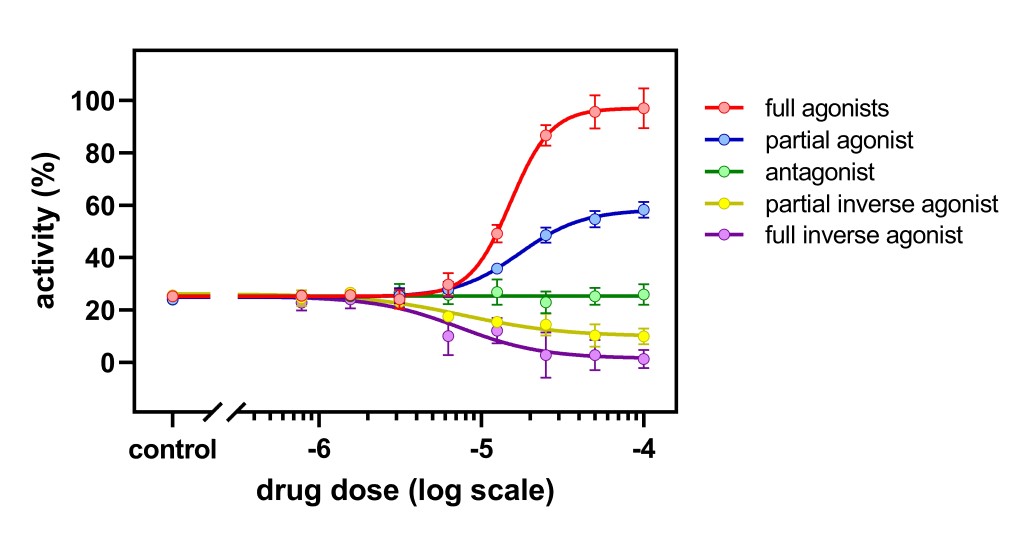
Figure 1. Dose-response curves depicting the activity profile of different ligand classes
Two determinants of drug receptor kinetics include affinity and intrinsic efficacy. Affinity is a drug’s ability to bind to a receptor. It depends on both the structures of the drug and the receptor. Medicinal chemists try to increase the selectivity of the chosen molecule by increasing affinity for the target receptor and minimize the affinity for off-target receptors. In the DPP4 example, increase selectivity to DPP4 and reduce affinity for DPP8/9. Often, such selectivity dialing is hard to achieve. You may see efficacy at the expense of some off target toxicity. Intrinsic activity is how a molecule modulates a receptor that yields a change in cellular activity. Recall the pioneering work of Nobel laureate Sir James Black with the drug propranolol, a beta-blocker that blocked the receptor for adrenaline; this was at the heart of the concept of intrinsic activity.
Another form of drug design uses secondary binding sites on receptors as the pharmacological targets. These are called allosteric sites, and compounds that bind to allosteric sites are called modulators. These will bind to allosteric sites on the receptor and modify the steric conformation of the primary orthosteric site of the target protein. Two new forms of modulators that you may come across are positive (modification that yields an increase in affinity of the ligand with orthosteric site) and negative (weakens the signal in this case).
Does your drug have an unambiguous response?
Now that you know the biology and the designer features of your drug, the next step is to understand whether you have a desirable effect or drug response. To measure drug effect, you either have a validated biomarker or surrogate endpoint, or you are screening a plethora of biomarkers in an exploratory manner. Drug effect could also be determined acutely or chronically. In the above example, if you inhibit DPP4 activity by 90% or greater, then you are likely to see a clinically meaningful fasting glucose effect. For neuroscience targets, you may be guided by receptor occupancy numbers as a benchmark number if your molecule exceeds a certain prespecified target.
If you have a drug for treating degenerative disease, even if you have an acute biomarker, you must justify the choice of that marker considering a longer-term clinical response. You may see an effect in short term studies not long term. Let’s take the example of rheumatoid arthritis. This is an autoimmune disease, and one can try to alleviate or reduce joint pain, or one can produce a disease modifying drug effect. In the latter, now you must rely on functional outcomes. Disease‐modifying antirheumatic drugs (DMARDs) slow progressive joint destruction. Methotrexate (MTX) is the most used DMARD, but several biologics are approved for RA with varied mechanisms of actions (e.g., anti‐ tumor necrosis factor, anti‐interleukin 1, anti‐IL‐6 receptor, B cell–depleting agents, Janus‐associated kinase inhibitor, etc.). Drug developers for RA are likely to use a proof of biology study to confirm an effect. However, the duration of effect is likely to be limited to a shorter trial duration. If one has a model relating the acute biomarkers to longer term outcome effect, then by changing one parameter, you can understand by how much the needle can move for longer durations. Trial simulations can predict the magnitude of the drug response over time.
A good example of target enrichment – a method to maximize the chance of discerning a treatment effect, is with tepotinib, an oral, highly selective MET tyrosine kinase inhibitor (Le et al., 2022). Recognizing that MET exon 14 (METex14) skipping is a key oncogenic driver in about 4% of non–small cell lung cancer cases, the developer hypothesized that such a patient population would benefit from targeted MET inhibitors. A phase 2 study in this enriched population yielded meaningful drug effect of objective response rate of 44.7% [95% confidence interval (CI): 36.7–53.0]. If you had selected a general non-small cell population, you may not have detected significant drug activity attributable to a driver mutation.
By ensuring you are designing the right clinical trial, enriching the right patient population, sample sizing for the right effect size, you are likely to reduce the uncertainty around drug response. Setting up robust decision criteria allows the drug developer to stop developing a drug with little probability of success of meeting the target product profile. The key is to be disciplined enough to walk away from the molecule or the mechanism if it fails to show an unambiguous effect.
To understand how biomarkers are helpful in drug development, read the whitepaper the author has written on this subject.
References
Le X et al. Tepotinib Efficacy and Safety in Patients with MET Exon 14 Skipping NSCLC: Outcomes in Patient Subgroups from the VISION Study with Relevance for Clinical Practice. Clin Cancer Res. 2022 Mar 15; 28(6): 1117–1126.
