


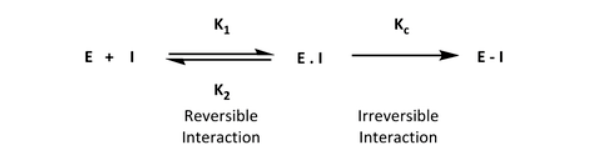
Traditional mechanisms of drug action rely on the reversible, noncovalent interaction of a ligand with its biological target. On the other hand, a targeted covalent inhibitor (TCI) drug is designed to form a covalent bond between an electrophile on the ligand and a nucleophilic center in the target protein (Figure 1). TCIs bind amino acids on the target protein by means of reactive groups in the drug, the so-called “warheads.” At first glance, one might think that a covalent bond is an uncommon and potentially harmful way to produce new medicines. However, recent interest in such covalent inhibitors has yielded drug hunting for many therapeutic targets. We will explore this further in this blog.
Aspirin, first available to patients in 1899, is the first truly covalent inhibitor, but that aspect wasn’t discovered or appreciated until much later. Now, covalency is targeted, and the covalent inhibitor approach is rapidly gaining acceptance as a valuable tool in drug discovery. Moreover, covalency is preferred as the next generation of modulators for previously intractable drug targets. The clinical success of ibrutinib, a Bruton tyrosine kinase inhibitor, for example in the treatment of mantle cell lymphomas, following its approval in 2013 spearheaded the interest in developing irreversible drug inhibitors (Figure 2). The other approved covalent drugs include acalabrutinib and zanubrutinib, which also inhibit Bruton tyrosine kinase.
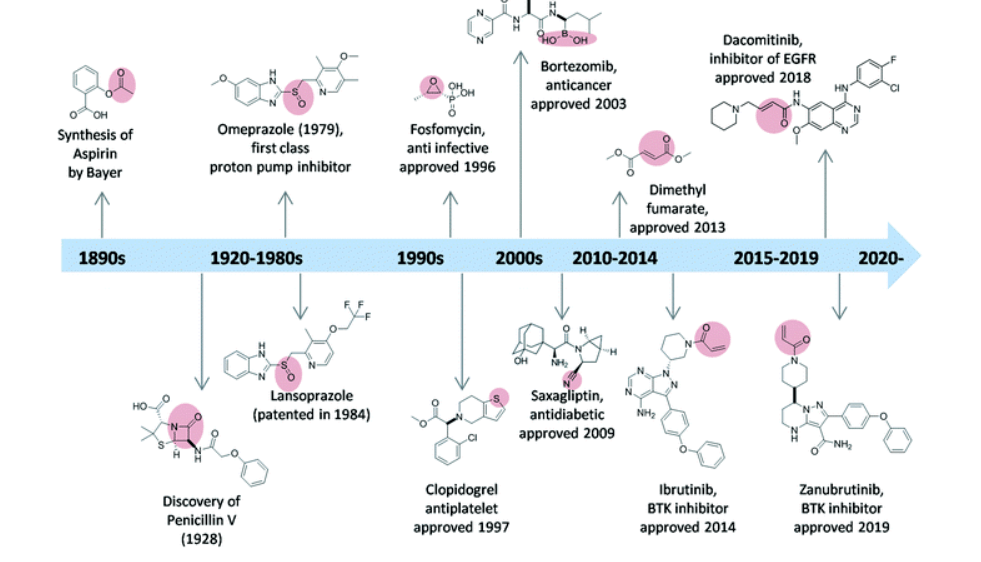
While the approved TCIs are mostly in the oncology and hepatitis areas, recent late-stage clinical trials are underway in a variety of autoimmune diseases with rilzabrutinib, evobrutinib, PF-06651600, and remibrutinib, among other therapeutic classes. In oncology, the recent accelerated approval of sotorasib demonstrates the applicability of the approach to a target previously labeled as “undruggable”. Sotorasib is a rat sarcoma (RAS) guanosine triphosphatase (GTPase) inhibitor that targets KRAS G12C by covalently binding to the cysteine of KRAS G12C. This covalent binding locks the protein in an inactive state in turn preventing downstream signaling without affecting wild-type KRAS. Overall, sotorasib reduces tumor cell signaling, inhibits cell growth, and promotes apoptosis in a subset of tumors harboring KRAS G12C, and is a good example of a precision medicine strategy.
So, why the sudden interest in TCIs? There are many theoretical benefits to the TCI approach. These include high potency (due to target blockade), low dose (due to high potency), and sustained duration of action (due to the long turnover time of the protein). Potency, dose, and durability are three strategic pillars of modern pharmacology, so it isn’t surprising that interest has blossomed in this area.
We share 3 main considerations in the development of TCIs.
Non-clinical drug metabolism and pharmacokinetics (DMPK)
DMPK screening during lead optimization of TCIs and subsequent clinical lead characterization should be tailored to the specific challenges associated with this class of small molecules.
From a DMPK perspective, non-clinical in vitro–in vivo extrapolation (IVIVE) for the prediction of clearance (CL), and subsequently human pharmacokinetic (PK) projection, may be challenging for TCIs due to their potential for conjugation with the cysteine moiety of glutathione (GSH) and potential non-specific binding.
A non-traditional excretion pathway for a subset of TCIs is via covalent binding to proteins, such as albumin, leading to relatively slow excretion in bound form following in vivo administration, as observed for afatinib and osimertinib (reviewed in Rioux and Nix, 2020; Stopfer et al., 2012). This phenomenon, in additional to chemical/enzymatic instability, may impact the accuracy of in vitro free fraction determination for TCIs which generally exhibit high plasma protein binding. Ex vivo or in vitro evaluation of covalent binding to plasma proteins allows for understanding of the fraction of the drug bound covalently to albumin and should be considered during development.
GSH conjugation may occur by non-enzymatic and/or GSH-S-transferase (GST)-mediated mechanisms in both the liver and extrahepatic tissues (Leung et al, 2017). GSH-related adducts are observed for most TCIs, albeit at a negligible level for some of them such as afatinib and ibrutinib (Scheers et al, 2015; Stopfer et al., 2012). Therefore, extrahepatic CL is a trait of TCIs. Consequently, data extrapolated from traditional matrices employed in in vitro CL assays such as recombinant human CYP enzymes, liver microsomes, liver S9 fractions, or hepatocyte incubations don’t typically represent in vivo CL. Consideration of whole blood stability (as an in vitro surrogate for GSH-conjugation in extrahepatic tissues) and blood binding has been proposed to improve CL IVIVE (Leung et al, 2017). Although challenging, achieving IVIVE is possible for TCIs as demonstrated with H3B-6545 in part via optimization of covalent warhead reactivity leading to reduction of extra-hepatic CL (Rioux et al, 2018). Despite recent advancement, gaps remain to establishing best practices for projecting human PK parameters for TCIs.
Clinical pharmacology
There are many aspects worth considering when planning the clinical pharmacology profiling of TCIs. They depend on the kinetics of the covalent bond formation. The relationship between drug concentration and the rate of covalent bond formation is critical. Ensuring the balance between a drug that has a selective and strong interaction with the target protein and the dose is important. This balance isn’t easy to achieve as there would be a fine tuning of the ratio of the maximal rate of inactivation (kinact) and potency (KI). These issues are further elaborated below.
Selectivity vs off-target reactivity
Balancing the reactivity of covalent warheads to reach high target occupancy versus potential for off-target reactivity is a key consideration in TCI development. In addition to potential safety risk, a subset of TCIs have been reported to be time-dependent inhibitors of at least one human CYP enzyme, highlighting the need for early characterization of drug-drug interaction (DDI) potential.
PK/PD decoupling
Covalent inhibition is time-dependent and prolonged exposure leads to increased target occupancy. That longer duration of action may decouple pharmacodynamics (PD) from plasma exposures (PK) for TCIs with moderate/high clearance rates. IC50 or EC50 values are not optimal for comparing covalent ligands. Potency of irreversible inhibitors is better expressed as kinact/KI, where kinact is the maximal rate of inactivation and KI is the reversible binding constant, which is a more labor-intensive assessment. In addition to kinetic constants, for PK/PD assessment, target protein synthesis and degradation rates must be taken into consideration. Note that a TCI approach is not suitable for targets with rapid turnover or requiring transient/partial inhibition. For targets with slower turnover rates, the potential for lower doses/less frequent dosing renders TCIs especially attractive. The long duration of action may decouple PD from PK for TCIs with high clearance rates.
Human absorption, metabolism, and elimination (hAME)
Despite the challenges associated with non-traditional pathways of excretion and potential for covalent binding to macromolecules, well designed human AME studies can achieve adequate recovery and allow determination of the route of elimination and major clearance pathways of TCIs (Rioux and Nix, 2020).
Drug Safety
The safety issues that were evident for early covalent inhibitors were idiosyncratic reactions because of reactive functionality of electrophiles in the molecules and/or their biotransformed products. As a result, the role of covalent adduct formation has been historically contextualized as an adverse effect of such drugs.
Given the inherent nature of TCIs, there is a theoretical potential of TCIs to produce immune-mediated adverse reactions triggered by modification of target or off-target proteins. While the drug and its protein target separately may not be immunogenic, the combination may be immunogenic by a process called haptenization where the covalently attached drug is the hapten (Figure 3). Such immunogenic drug–protein adducts might trigger idiosyncratic reactions, often weeks or months after initiation of therapy. Although the approved TCIs have passed regulatory muster in terms of safety and efficacy, the safety and long term risks of TCIs would benefit from additional investigation and research.
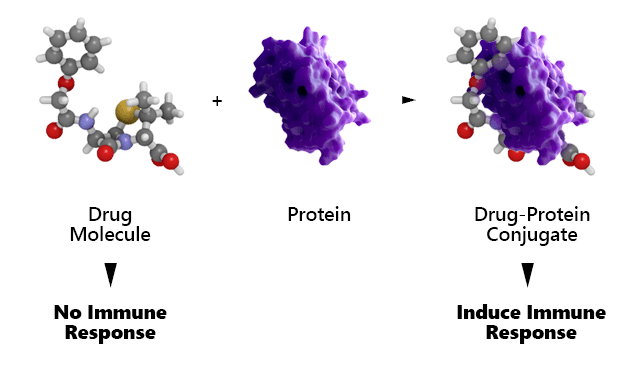
Increasing the selectivity in target engagement together with high potency at low doses and dialing out reactive metabolite formation from cytochrome P450-mediated reactions may mitigate against these immunogenic side effects. In this regard, ensuring TCIs with optimal balance of kinact/KI from a drug discovery perspective is essential to safeguard against long term safety issues.
Conclusions
When rationally designed, TCIs have the potential of generating highly potent, selective, and safe therapeutics for a wide array of diseases. There are critical DMPK and clinical pharmacology issues that need to be rationalized, as highlighted in this blog. To learn more about how our clinical pharmacologists can support your drug program, visit this webpage.
References
- Leung et al (2017). Clearance prediction of targeted covalent inhibitors by in vitro-in vivo extrapolation of hepatic and extrahepatic clearance mechanisms. Drug Metab Dispos 45:1–7.
- Rioux N, and Nix, D. Characterization of Human AME for Targeted Covalent Inhibitors (TCIs): Review of FDA Oncology Approvals, AAPS PharmSci360, 2020, Virtual meeting, Special Poster Collection, Poster 879921.
- Rioux N, Smith S, Korpal M, O’Shea M, Prajapati S, Zheng GZ, Warmuth M, Smith PG. (2019) Nonclinical pharmacokinetics and in vitro metabolism of H3B-6545, a novel selective ERα covalent antagonist (SERCA). Cancer Chemother Pharmacol. 83:151-160.
- Scheers E, Leclercq L, de Jong J, et al. (2014). Absorption, metabolism, and excretion of oral 14C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos 43:289–97.
- Stopfer P, Marzin K, Narjes H, et al. (2012). Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother Pharmacol 69:1051–61.
- Sutanto F, Konstantinidou M, Dömling A. Covalent inhibitors: A rational approach to drug discovery. RSC Med Chem. 2020;11(8):876-884. http://dx.doi.org/10.1039/D0MD00154F. doi: 10.1039/D0MD00154F.

