



Investigational New Drug Applications (INDs) and Clinical Trial Applications (CTAs) are regulatory submissions needed for the initiation of clinical drug trials, and these applications have commonalities and differences throughout the world. If you’re initiating clinical trials in multiple countries, then planning for these different submissions is critical to the efficiency of your drug program. Many drug developers struggle to get this planning right and stay on top of changing regulations. That’s why this blog will compare the authoring of documents included in these submission types to the United States (US), European Union (EU), United Kingdom (UK), and Canada (Table 1), along with insights and recent regulatory updates.
An IND is used to initiate clinical trials in the US and is submitted to the Food and Drug Administration in electronic Common Technical Document (eCTD) format (Figure 1). There are limited Module 2 summaries depending on the product history. All underlying source reports, including Standard for Exchange of Nonclinical Data (SEND) datasets for applicable nonclinical studies, must be included (learn more about SEND in this blog). Luckily, once submitted for a specific drug and indication, all future studies, documents, and data for that drug and indication are submitted as additions to that same IND.
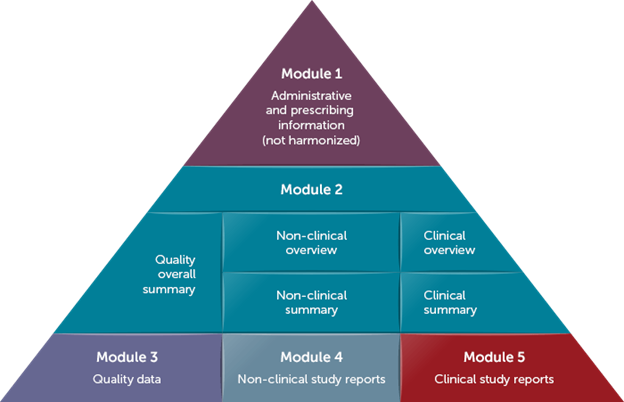
A CTA is used for initiating clinical trials throughout most of the rest of the world. Most countries accept chemistry, manufacturing, and controls (CMC), nonclinical, and clinical information summarized in an Investigational Medicinal Product Dossier (IMPD). As of 31 January 2023, the EU only accepts CTAs that are submitted via the Clinical Trial Information System (CTIS; learn more about this clinical trial database in this blog). A CTIS submission includes the IMPD and cannot accommodate eCTD sections or structure. An added benefit of CTIS is that applications for up to 30 countries can be requested with the same documentation. Also, healthcare providers and patients can search public portions of the CTIS database for information.
There haven’t been any changes to CTA submission requirements for either the UK or Canada. The CMC section needs to be complete (equivalent quantity and level of information as in Module 3 in eCTD), and source reports are not necessary. The nonclinical and clinical sections can be very brief, high-level summaries that largely cross-reference the Investigator Brochure (IB). All this information is submitted each time the Sponsor wishes to initiate a new clinical trial, regardless of whether a CTA has already been submitted for a specific drug and indication. However, subsequent studies can cross-refer to the initial CTA, especially for the same CMC material.
For all 4 geographic areas (US, UK, Canada, and EU) discussed in this blog, the IB, Protocol, and Informed Consent Form are submitted in full in each region.
It is important to carefully plan for submission to multiple countries. The decision on which country to submit to first will have a waterfall effect on subsequent decisions such as eCTD, IMPD, or both formats (if submitting an IND and CTA) as well as how to most efficiently develop the CMC, nonclinical, and clinical content. For example, if a CTA will be submitted first, then the nonclinical information will be within the IB, while an IND will require the completion of the eCTD Module 2 summaries. Conversion from a CTA to an IND is more complex and requires more time and effort than the reverse.
Certara can assist you with your submission needs and can tailor your application to your unique clinical development program.
| Table 1 Comparison of Initial Regulatory Applications Across Geographical Regions | ||||
| Geographic Region | US | EU | UK | Canada |
| Submission Type | IND | CTA | CTA | CTA |
| Structure | eCTD | IMPD | IMPD | eCTD or IMPDa |
| CMC | Full eCTD Module 3 but can often skip creating eCTD Module 2.3 | Full IMPD Section 2 | Full IMPD Section 2 | Full eCTD Modules 2.3 and 3 or IMPD Section 2 |
| Nonclinical | Full eCTD Modules 2.4, 2.6s, and 4 (including SEND datasets)b | IMPD Section 3, including only a brief summary cross-referencing the IBc; no study reports or SEND datasets | Not includedd | Not includedd |
| Clinical | eCTD Modules 2.5 and 2.7 as well as the proposed Protocol and ICF in the appropriate Module 5; Any study reports and appendices must also be included in the appropriate Module 5 | IMPD Section 4 includes a brief summary cross-referencing the IB as well as the proposed Protocol and ICF; no study reports | Not includede | Not includede |
| Submission Mechanism | FDA ESG | CTIS | IRAS Portal | CESG through FDA ESG for eCTD submissions or by mail for IMPD submissions |
| CESG=Common Electronic Submissions Gateway; CTA=Clinical Trial Application; CTIS=Clinical Trial Information System; eCTD=electronic Common Technical Document; ESG= Electronic Submissions Gateway; EU=European Union; IB=Investigator’s Brochure; ICF=Informed Consent Form; IND=Investigational New Drug Application; IRAS=Integrated Research Application System; SEND= Standard for Exchange of Nonclinical Data; UK=United Kingdon; US=United States. a Noncommercial CTA are often submitted as IMPD format. b In some cases, some of the Module 2 summaries can be omitted and tailored for the IND. c No study reports or SEND datasets should be included. d This is considered covered by the IB, so nothing additional needs to be submitted. e This is considered covered by the IB, Protocol, and ICF, so nothing additional needs to be submitted. |
||||
Get in touch with our IND and CTA applications experts
Certara offers expert support for your submission needs, tailoring each application to fit your unique clinical development program. With our advanced regulatory submission solutions, we simplify complex processes, ensure compliance, and help accelerate your drug development timeline with confidence.
