



The end of 2018 ushered in a flurry of new regulatory guidance and sponsor enthusiasm on real-world evidence (RWE) and its adoption in the drug development process. While the collection of real-world data (RWD) and use of RWE is not new, they are now poised to have a profound impact on our industry. Today, it is common practice for regulators to use RWE to monitor post-market safety and to make regulatory decisions. And increasingly, sponsors have been leveraging RWE to support both clinical trial design and observational studies to generate treatment approaches. Likewise, healthcare systems are collecting and using RWE to substantiate coverage decisions.
FDA Publishes its RWE Framework
While clinical trial evidence remains the gold standard for evaluating treatment efficacy, there is increasing interest and potential for leveraging RWD to inform healthcare decision-making. Both the 21st Century Cures Act and the PDUFA VI required the FDA to create a framework for addressing how RWE can be used to better support regulatory decisions. That framework, published at the end of 2018, begins with some key definitions:
- Real-World Data (RWD) are data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources.
- Real-World Evidence (RWE) is the clinical evidence about the usage and potential benefits or risks of a medical product derived from analysis of RWD.
- Examples of RWD include data derived from electronic health records (EHRs); medical claims and billing data; data from product and disease registries; patient-generated data, including from in-home-use settings; and data gathered from other sources that can inform on health status, such as mobile devices. RWD sources (e.g., registries, collections of EHRs, administrative and medical claims databases) can be used for data collection and, in certain cases, to develop analysis infrastructure to support many types of study designs to develop RWE, including, but not limited to, randomized trials (e.g., large simple trials, pragmatic clinical trials) and observational studies (prospective or retrospective).
According to Janet Woodcock, MD and Director of FDA CDER, “FDA will work with its stakeholders to understand how RWE can best be used to increase the efficiency of clinical research and answer questions that may not have been answered in the trials that led to the drug approval, for example how a drug works in populations that weren’t studied prior to approval.”
Specifically, FDA’s RWE Program will evaluate the potential use of RWE to support changes to labeling about drug product effectiveness, including adding or modifying an indication, such as a change in dose, dose regimen, or route of administration; adding a new population; or adding comparative effectiveness or safety information. The framework will include these considerations:
- Whether the RWD are fit for use
- Whether the RWE study design can provide adequate scientific evidence to help answer the regulatory question
- Whether the study conduct meets FDA regulatory requirements (e.g., for study monitoring and data collection)
Pilot projects are underway, and the agency is seeking additional sponsors for such partnerships. This provides a new and exciting opportunity for Pharma and its partners to explore new and innovative ways to use RWE to fast-track products to market; cutting the cost of large phase III trials and massively reducing the waiting time for patients to receive life-changing new therapies.
EMA’s “Regulatory Science to 2025” Rallies Behind RWE
The EMA just published its ‘Strategic Reflection: Regulatory Science to 2025’ document. Aligned with the FDA and other global regulators, the EMA views RWE alongside cell-based therapies, genomics-based diagnostics, drug-device combinations, novel clinical trial design, predictive toxicology, modeling & simulation, ‘big data,’ and artificial intelligence as transformative research endeavors.
To that end, EMA is seeking to:
- create a sustainable, quality assured, flexible framework delivering rapid access to and analysis of representative, longitudinal RWD throughout a product’s lifecycle;
- develop a capacity that will enable EMA to rapidly and securely access and analyze large amounts of healthcare data;
- accelerate the implementation of a learning regulatory system based on health economics and outcomes research (HEOR) and other clinical care data;
The agency recognizes the benefit of using RWD to generate complementary evidence across the product life cycle and is committed to promote the use of high quality RWD in decision-making. EMA is further offering consultations in parallel with the European network of health technology assessment bodies (EUnetHTA).
National health systems have long been interested in RWE partnerships. A recent engagement with the French National Authority for Health (Haute Autorité de Santé; HAS) presents a powerful example. The RWE study involved 600+ patients over 6 centers for a conditional reimbursement scheme in chronic obstructive pulmonary disease (COPD). Over an 18 month timeframe, the therapy was shown to significantly reduce the number of hospitalizations and, therefore, remained fully reimbursed. Understanding these opportunities and choosing the right framework for your evidence approach is where expert guidance makes the difference.
Advancing RWE with Certara
In 2018, Certara acquired Analytica Laser, a leading provider of market access and evidence services. Behind the company’s quantitative solutions stands an industry-leading team focused on RWE strategies and data analysis for commercial and scientific applications. Driven by the growing value of RWD in generating regulatory-meaningful evidence, experts lead projects to:
- identify opportunities across the clinical development cycle where RWE can answer critical clinical and commercial questions;
- evaluate healthcare technology in the context of public health needs and design RWE study protocols that meet the need of future payers;
- assess and collect relevant data (sources) and aggregate that data in a manner relevant to health systems’ requirements;
- perform outcomes research and surveys;
- define patient-reported outcomes;
- conduct burden of illness studies;
- perform retrospective data analysis and natural history patient population studies;
- study relative comparative effectiveness in real-world conditions.
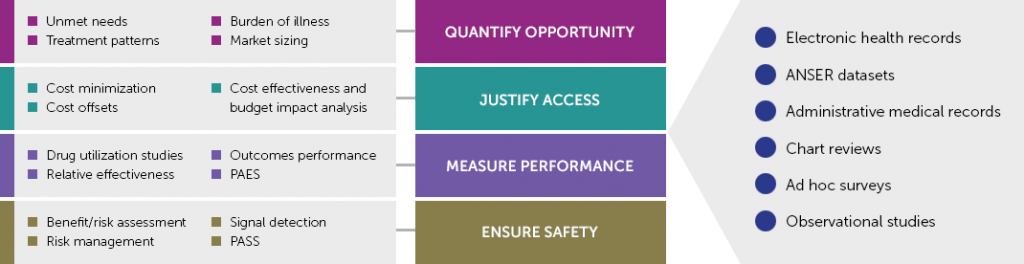
Our teams are proud to be the first successful non-academic applicant to get a positive response towards a research inquiry using the new French health system’s ‘SNDS’ patient database of over 50 million lives. In 2018, we partnered with many industry clients to pioneer accessing and investigating the new SNDS data. Combined with our own ANSER Real World Data Sets and other databases in Europe, this offers a combined population of more than 100 million lives in Europe.
Patient-Reported Outcomes (PROs) as a Benchmark of RWD Excellence
Patient data systematically recorded from routine clinical settings (such as PROs) are one of the key enablers of regulatory acceptance of real-world evidence. However, even strong proponents of drawing on RWD acknowledge that ‘the real world’ can be messy. The data our research is likely to draw on are often as fragmented, unstructured, and multifaceted as the settings they emerge from. More than ever, experience in closing the so-called efficacy-to-evidence gap is required to formulate evidence strategies in line with the value proposition of novel technologies.
The strong benefits of PROs to the product development strategy rest on high quality scales that can address the target audience’s constructs of interest. Our experts help customers choose the most appropriate tools for the research context: health-related quality of life (HRQoL), satisfaction with treatment, adherence, or symptom measures. We can perform cultural adaptations of PROs across different countries, and we design and perform validation studies to assess their psychometric properties (reliability, validity, and sensitivity to change). For cases where disease areas lack robust tools, Certara was able to develop new, reliable ones. We apply Classic Theory, Rasch Model and Item Response Theory in developing and validating PROs. We also have experience in developing Computer Adaptive Test based on IRT.
Certara’s Responsibilities as part of the IMI ‘Get Real’ Initiative
The Innovative Medicines Initiative (IMI) is Europe’s largest public-private partnership to improve the drug development process. IMI consists of pharmaceutical companies, academia, HTA agencies and regulators (e.g., NICE, HAS, EMA and ZIN). The GetReal Initiative is focused on the adoption of tools, methodologies, and best practices for increasing the quality of RWE generation in medicines development and regulatory/HRA processes across Europe. An active member of this consortium, Certara is specifically involved in the statistical approaches for pragmatic trials and development of best practice recommendations, along with the use of both network meta-analysis (NMA) and multi-criteria decision analysis (MCDA) for assessing the relative effectiveness of drugs.