



Next generation therapeutics
Oligonucleotide drugs are unique among therapeutic modalities in that they don’t engage the disease modulating proteins directly but instead target their encoding mRNA (read more about how “oligos” work in this blog). The benefits of this approach are manifold. First, from the synthetic chemistry point of view, different drugs can be assembled in a standardized fashion from the same nucleotide building blocks with only the drug sequence varying according to the target protein mRNA. This allows the chemistry, manufacturing, and controls (CMC) process to be optimized across the development portfolio while the chemical properties of resulting oligonucleotide therapeutics are highly conserved from one drug to another, thus vastly accelerating the bench to bedside transition.
Moreover, the nature of the disease-modulating protein itself plays little to no role at all. It doesn’t matter whether this protein is intra- or extracellular, soluble, or membrane-bound or even if it has any stable structure altogether, the latter being the common unstated requirement both for small and large-molecule drugs. Finally, future therapeutic interference by oligonucleotides can be even more subtle through gene expression regulation, editing, or non-coding RNA inhibition, etc. Accordingly, it’s no surprise that the number of oligonucleotide drugs approved is growing rapidly, reaching fifteen now since Fomiversen, the first of them, was made available for Cytomegalovirus Retinitis in 1998 (Figure 1). The oligo market is anticipated to follow a steady annual growth rate of 13% until 2030.[1] [2]
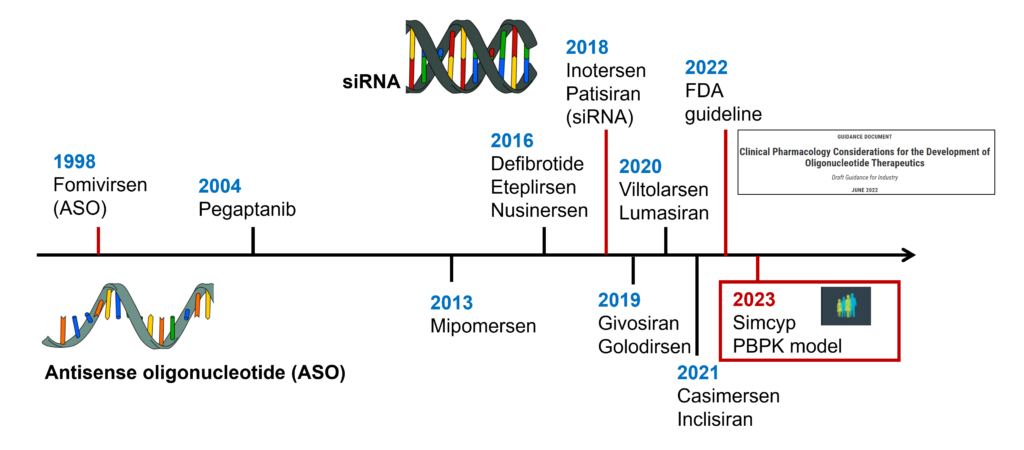
Adaptation of oligonucleotide therapeutics to specific roles
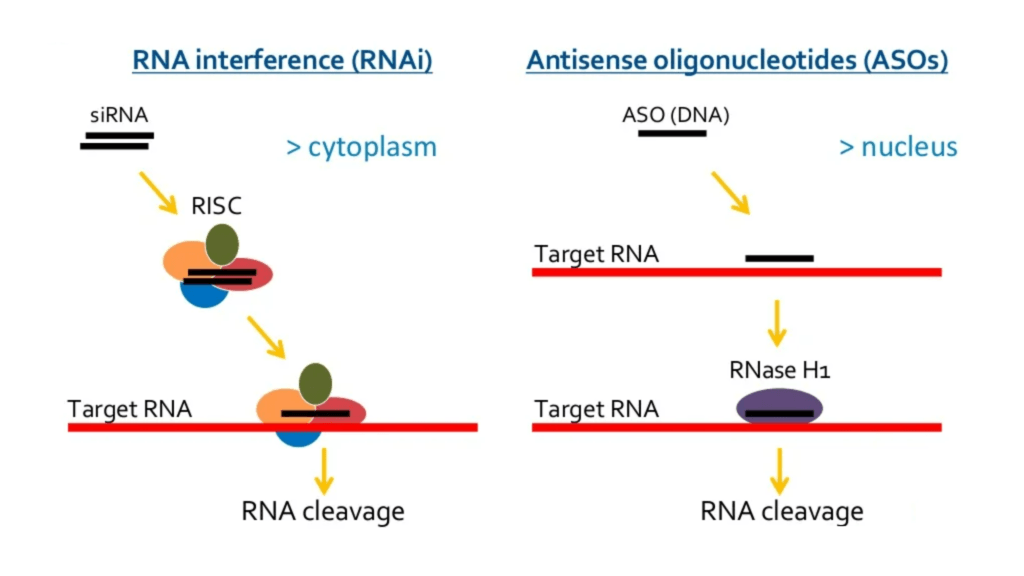
Currently, the two most common types of oligonucleotide drugs are single stranded antisense oligonucleotides (ASO) and double-stranded small interfering RNAs (siRNA), which have both similarities and differences. On one hand, while both target the mRNA of proteins of interest for degradation, their mechanisms of action are different (Figure 2), with ASOs relying on RNase H-mediated cleavage of the mRNA-DNA duplex and siRNA on the activity of RNA induced silencing complex (RISC). The intracellular site of action, with the target mostly found in the cytoplasm, reveals the greatest challenge impeding the deployment of therapeutic oligonucleotides to date.
The physicochemical properties of therapeutic oligonucleotides break nearly all of the five rules of Lipinski. [3], being large, typically 7-12 kDa, highly charged, and capable of forming dozens of hydrogen bonds. As such, therapeutic oligonucleotides cannot reach the site of action directly by diffusion across the cell membrane. Instead, scavenger receptor mediated uptake is the main route of cellular uptake as a substantial fraction of the dose ends up in the liver [4], which can be further facilitated by asioaloglycoprotein receptor (ASGPR) mediated uptake for GalNAc-modified oligos.[5]
The challenges oligonucleotide therapies must overcome
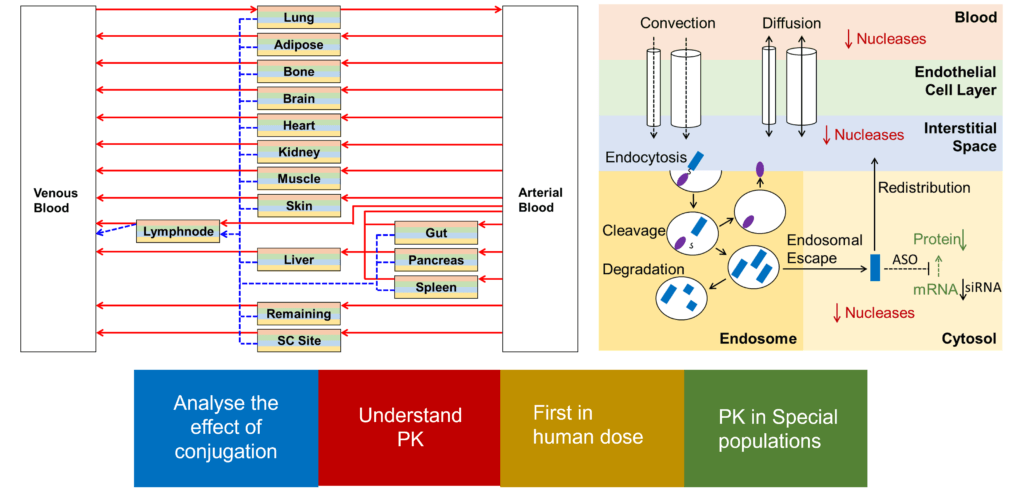
Firstly, one needs to understand and develop a predictive mechanistic model that describes the tissue distribution and uptake kinetics of oligonucleotides irrespective if they are unmodified or conjugated to a targeting group like GalNAc, a peptide, or even an antibody. Secondly, there is a need to quantitatively understand and describe the process of intracellular trafficking of oligonucleotide therapeutics following receptor-mediated uptake, as the drug moves from endosomes and lysosomes to cytoplasm. Once these are met and incorporated into a mathematical model, one can expect to use PBPK modeling (Figure 3) to:
- predict and analyze the concentration at the site of action of both target tissues and other organs
- quantify the contributions from parallel routes of cellular uptake and any impact from targeting moieties
- account for the degradation of oligonucleotides in tissues and elimination of both degradation products as well as intact drug
- quantify cellular escape of the drug into circulation
- predict first-in-human dose
- compare different formulations
- support the translation to special populations
To conclude, under the guidance of the Simcyp Consortium members, we are actively working on a novel oligonucleotide PBPK model that will be implemented into the Simcyp Simulator V23. To learn more about our oligonucleotide PBPK model, please contact us.
References
1. Migliorati, J.M., et al., Absorption, distribution, metabolism, and excretion of FDA-approved antisense oligonucleotide drugs. Drug Metabolism and Disposition, 2022: p. DMD-MR-2021-000417.
2. Biopharma-PEG. Oligonucleotide Drugs: Current Status and Challenges. 2020; Available from: https://www.biochempeg.com/article/124.html.
3. Lipinski, C.A., et al., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 2001. 46(1-3): p. 3-26.
4. Miller, C.M., et al., Stabilin-1 and Stabilin-2 are specific receptors for the cellular internalization of phosphorothioate-modified antisense oligonucleotides (ASOs) in the liver. Nucleic acids research, 2016. 44(6): p. 2782-2794.
5. Jeon, J.Y., V.S. Ayyar, and A. Mitra, Pharmacokinetic and Pharmacodynamic Modeling of siRNA Therapeutics – a Minireview. Pharmaceutical Research, 2022. 39(8): p. 1749-1759.

