



This is the first of three blog posts discussing best practices for developing oligonucleotides.
Interest in oligonucleotide therapies, which are composed of short strands of DNA or RNA, is growing rapidly; there are at least 135 of them currently in development.1 Underscoring their proliferation, a PubMed search for “oligonucleotide therapeutics” generated more than 35,000 hits. Furthermore, it is estimated that by 2024, the global nucleic acid therapeutics market will reach a value of more than US $7 billion.2
Historical Context
Nucleic acids, DNA and RNA, were first discovered in 1868, but most of the research that went into founding the oligonucleotide therapeutic class was done in the 20th century (see Figure 1). It builds upon biology’s central dogma – the idea that DNA is transcribed to RNA, which is translated to protein.
Scientists realized that if they could create oligonucleotides that interfere with the expression of RNA linked to disease-causing proteins, those entities could serve as therapeutics.
In this series of blog posts, we will focus on two types of oligonucleotides: antisense oligonucleotides (ASOs), which are single-stranded DNA snippets, and small interfering RNAs (siRNA), which are double-stranded RNA molecules.
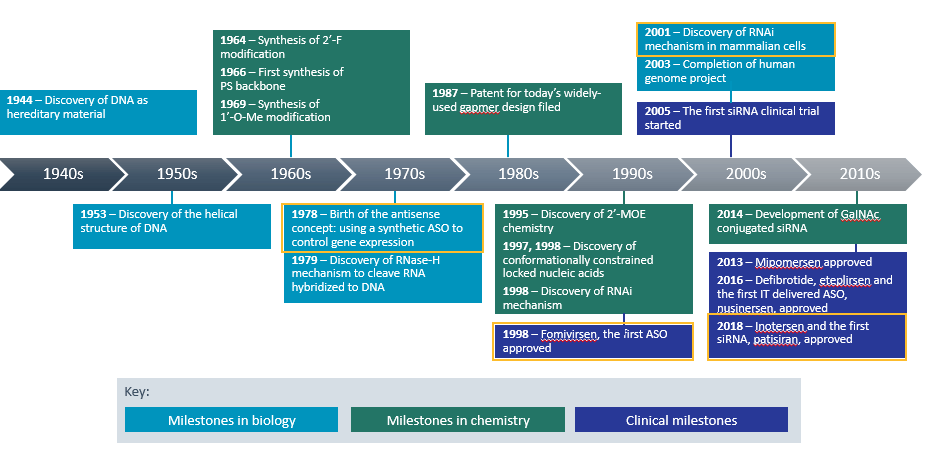
Clinical and Translational Science, Volume: 12, Issue: 2, Pages: 98-112, First published: 01 February 2019, DOI: (10.1111/cts.12624)
Scientists first conceived the idea of using a synthetic ASO to control gene expression in 1978. Twenty years later, the FDA gave its first ASO approval to fomivirsen sodium intravitreal injection for the local treatment of cytomegalovirus retinitis in patients with acquired immunodeficiency syndrome (AIDS).3
Then in 2001, came the discovery of the RNA interference (RNAi) mechanism. In 2018, this led to the approval of patisiran, the first siRNA therapeutic. Patisiran infusion is indicated for the treatment of peripheral nerve disease (polyneuropathy) caused by hereditary transthyretin-mediated amyloidosis (hATTR) in adult patients.5
To help increase their targeting to specific tissues, drug developers sometimes conjugate oligonucleotides to ligands. The best-known example is multivalent N-acetylgalactosamine (GalNAc), a high-affinity ligand that binds specifically to asialoglycoprotein receptors (ASGPRs), which are highly expressed on the surface of hepatocytes.
Comparisons with Other Drug Modalities
How do oligonucleotides compare with small molecule and biologic drugs? Oligonucleotides are intermediate in size; they are bigger than small molecules, but smaller than biologics, such as peptides and proteins.
Oligonucleotides are also highly soluble, and because they have a sugar phosphate backbone, they are polyanionic, highly negatively charged.
In terms of their pharmacokinetic characteristics, oligonucleotides are highly distributed to the liver, kidneys, fat cells, lymph nodes, spleen, and bone marrow, but not the brain. This is important for central nervous system (CNS) drug developers – oligonucleotides tend to be too large and too negatively charged to cross the blood-brain barrier.
While small molecule drugs can often be administered orally, oligonucleotides and biologics have historically been restricted to subcutaneous and intravenous administration. Furthermore, to target a CNS indication, oligonucleotides have traditionally needed to be administered through more invasive methods, such as an intrathecal injection, where they are introduced directly into the cerebrospinal fluid.
However, recent research3 shows that absorption enhancers, such as sodium caprate, may serve to enhance the intestinal permeability of oligonucleotides and facilitate oral administration,4 which would greatly benefit patients.
Oligonucleotide metabolism is mediated by exo- and endo-nucleases. Then, they are eliminated by the liver and kidneys.
Toxicity Considerations
One of the challenges with small molecule drug development is off-target toxicity. However, oligonucleotides are rationally designed against their mRNA targets, so off-target toxicity tends not to be an issue.
Immunogenicity, which occurs when a patient’s immune system mounts an immune response to an administered drug, is a well-known challenge with some biologics. ASOs, which represent novel structures, also have the potential to trigger immunogenicity – a possible problem of which drug developers should be aware.
Mechanisms of Action
Both ASO and siRNA therapeutics work by mediating degradation of the target mRNA and blocking expression of the protein target, but they use different mechanisms.
An ASO is synthetic, single-stranded DNA, which is complementary to the mRNA target. The ASO binds its target mRNA through Watson-Crick base pairing, and then downregulates the molecular target usually by inducing RNase H endonuclease activity. This cleaves the RNA-DNA heteroduplex, resulting in significant reduction in target gene translation.
In contrast, siRNA is a double-stranded RNA entity. It enters the cell and is cleaved by the Dicer endoribonuclease, which allows it to be incorporated into other proteins to form the RNA-Induced Silencing Complex (RISC). This unwinds the double-stranded complex to form single-stranded siRNA, which binds to its target mRNA. The cell then recognizes the mRNA as abnormal and begins to degrade it. As translation of that mRNA into amino acids and then protein does not occur, the gene which encodes it is effectively silenced.
Formulations
Most ASOs can be administered in simple formulations, such as saline, but siRNAs and modified mRNAs generally require a protective formulation, such as a lipid nanoparticle (LNP) delivery system. Within the LNP, the negatively charged siRNA is bound electrostatically to positively charged, cationic lipids. That is the form in which these therapeutics are typically administered.
Conclusion
Oligonucleotide therapies represent an elegant, targeted, successful approach to developing new drugs for genetic diseases in multiple therapeutic categories. In the remaining two blog posts in this series, we will discuss pre-clinical, translational, and clinical pharmacology considerations for oligonucleotide drug developers.
To learn more about best practices for oligonucleotide therapeutics development, please watch this webinar:
Dr. Minton is the Director of Content Strategy at Certara. She earned her Doctorate in Pharmacology from the UNC Chapel Hill.
References
- https://www.biospace.com/article/oligonucleotide-therapeutics-market-increased-research-and-development-activities-in-the-field-to-shoot-up-demand/
- https://www.globenewswire.com/news-release/2020/10/20/2110809/0/en/Global-7-23-Bn-Nucleic-Acid-Therapeutics-Oligonucleotide-Markets-2020-2024-Insights-Forecast-with-Potential-Impact-of-COVID-19.html
- Gennemark. Oral antisense oligonucleotides and PKPD considerations. DMDG 2021. https://www.dmdg.org/events/20210119/programme.asp
- FDA approval of fomivirsen. https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/20961_Vitravene.cfm
- FDA approval of patisiran.
https://www.fda.gov/news-events/press-announcements/fda-approves-first-its-kind-targeted-rna-based-therapy-treat-rare-disease
