



October 7, 2025
Alzheimer’s disease (AD) is one of the most common forms of neurodegenerative dementia in the United States. In fact, the Alzheimer’s Association predicts that by the year 2050, the number of people age 65 and older with Alzheimer’s dementia is expected to double, to comprise 12.4 million patients. AD is caused by a complex dysregulation of neurobiological networks and cellular functions. Such impairment causes neuronal and synaptic loss leading to memory dysfunction. Until Biogen’s approval of aducanumab, available treatments were largely symptomatic to address cognitive dysfunction. These included cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) and an N-methyl-D-aspartate antagonist (memantine).
One widely debated mechanism of AD that has been extensively researched by academic laboratories and industry alike is the “amyloid hypothesis.” In simple terms, the amyloid hypothesis posits that the core reason for cognitive decline is due to amyloid beta (ABeta) deposits in the brain. However, healthy people with no AD-like symptoms also have amyloid deposits in their brains. In addition, there is no meaningful relationship between degree of amyloid deposits and degree of cognitive decline, which makes drug traction nearly impossible to assess. Refer to the insightful commentary by Hardy and Selkoe in their seminal 2002 Science paper on this subject for more details (1).
Many investigational small molecule drugs targeted at the amyloid hypothesis including gamma and beta secretase inhibitors have fallen by the wayside. There are, of course, other, less understood mechanisms including the tau mechanism wherein these proteins become hyperphosphorylated insoluble aggregates called neurofibrillary tangles. For this blog, we will consider amyloid-based mechanisms.
The recent approval of aducanumab represents the first truly disease-modifying treatment of AD. Aducanumab is a human immunoglobulin G1 (IgG1) monoclonal antibody against the aggregated forms of amyloid-β pathway. This mechanism is purported to accelerate the clearance of β-amyloid from the brain through microglia-mediated phagocytosis.
What makes AD so difficult to treat?
The pathological cascade underlying deterioration in AD is one issue, but for truly disease modifying therapies, it also concerns mechanisms of cognitive decline. A third confounder relates to difficulty translating between animal models and humans, and from healthy subjects to AD patients. A fourth is the subjective nature of cognitive assessments, such as the Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog), Cambridge Neuropsychological Test Automated Battery (CANTAB) and the Mini-Mental State Examination (MMSE), and the difficulties surrounding signal identification.
In this blog, we propose using an integrated model-informed drug development strategy for Alzheimer’s disease to maximize the “learn and confirm” nature of clinical trials involving new drugs. These key considerations are as follows.
Ensure optimal DMPK properties
Ensuring optimal drug metabolism and pharmacokinetic (PK) configuration is essential for small molecule investigational drugs as well as biodistribution capabilities for large molecules, as the target site of action or biophase compartment is often within the central nervous system, requiring molecules to cross the blood-brain barrier (BBB). Thus, crossing the BBB is an important absorption, distribution, metabolism, excretion (ADME) characteristic that CNS-targeting drugs should have. Generally, these compounds should have CNS “likeness” properties such as molecular weight < 400, moderate lipophilicity (clogP < 3), and the number of hydrogen bond donors and acceptors < 4 and < 8, respectively, for enabling a compound to cross BBB (2).
Designing a molecule that crosses the BBB isn’t easy. There are two components for this transport, either passive (diffusion mechanism) or an active transport mechanism. Several assays can be used and interpreted alone or together to make a rational decision on the ADME behavior of a compound that is being developed for a CNS target. Permeability across the BBB can be determined through either in situ brain perfusion technique or in vitro cell culture such as primary culture porcine brain endothelial cells or immortalized cells. The MDCK cell line (dog kidney epithelial cells) transfected with MDR1 gene has been widely used in the pharmaceutical industry for predicting BBB permeability as well as determining P-glycoprotein efflux potential.
In addition, determining the in vivo brain/plasma ratio in non-clinical species, which relates to the extent of brain penetration, is an industry best practice. Balancing the rate and extent of a drug’s brain penetration is an important factor in designing optimal CNS PK properties (2). Delivering large molecule therapeutics across the BBB has been a major obstacle to successful drug development in neurodegenerative diseases and is critical to enabling effective treatments. Not only must the molecule reach the brain, but it must also engage with the pharmacological target to ensure a rapid onset of action and durable response. For these reasons, a brain penetrable molecule design is critical with an acceptably long residence time in the biophase compartment. A good example for brain transport vehicles is illustrated by Denali Therapeutics. Denali’s website indicates transferrin receptor based large molecule transport vehicle as a platform to engineer BBB access.
Regardless of small or large molecules, ensuring site of action access as well as measuring drug concentrations in the brain or surrogate cerebrospinal fluid compartment can give valuable insight to biophase concentrations. In combination with biomarker measurements in the same compartment, they can assist in understanding dose/concentration relationships as well as exposure/response relationships.
Build credible clinical pharmacology and translational tools
A robust translational strategy is a cornerstone for any neurodegenerative drug development program. There are many options to choose from.
Early phase programs typically include biomarkers of proximal target engagement and a host of neurobiological markers. Due to the small sample size inherent in phase 1 trials, there is considerable analytical noise precluding quantitative relationships. If tractable, receptor occupancy studies can assist with identifying the extent of occupancy that would be necessary to trigger beneficial pharmacology. Concentration/RO relationships can then assist with the selection of EC50 values that could guide dose selection in terms of identifying clinically relevant concentrations.
Another example of a translational tool is the scopolamine challenge model. Specific to the study of cholinergic drugs, the scopolamine challenge study is often done both preclinically and in humans and can provide translatability between animals and humans. The principle of this study is that scopolamine is a muscarinic antagonist that impairs cognitive function, and that the extent of the pharmacodynamic effect of investigational drugs could be assessed by the degree of reversal of this effect by scopolamine. This effect is measured using the Groton Maze Learning Test (GMLT) given as part of the CogState neuropsychological test battery. While extensively applied, the model is fraught by methodological concerns including the safety of administering scopolamine.
Because the drug effect on the body is often constrained by longitudinal analysis, translatability of preclinical to clinical investigation is often fraught by methodological issues. Take plaque regression for instance. One could easily design an experiment looking at plaque burden in a long-term animal study. And while one sees meaningful drug-induced reductions in plaque in animals, these findings are often not readily translated into early phase clinical trials that are limited in duration. Including a meaningful translational study in the clinic often aids understanding dose and exposure/response relationships. One good example of such a translational approach is a small Phase 1b study (PRIME) performed with varying doses of aducanumab (3). The authors used an 18F-labeled florbetapir radioligand to bind selectively to Abeta which allows the investigators to directly image the plaques in AD patients using positron emission tomography (PET). This approach allows quantifying changes in plaque burden as a function of time with an investigational drug. Their study revealed both time and dose-dependent decreases in amyloid PET standardized uptake value ratio (SUVR) with aducanumab, a finding that prominently featured in the FDA’s assessment of effectiveness in addition to the two phase 3 trials (4).
Quantitative strategies to maximize model-informed drug development decisions
A plethora of modeling strategies have been applied for AD drug development. These range from empirical to mechanistic modeling, but also physiologically-based PK modeling and disease progression modeling. More sophisticated quantitative systems pharmacology (QSP) modeling has also been used widely. Ensuring a robust MIDD strategy will ensure exposure and/or dose/biomarker/disease response relationships are fully leveraged and interpreted.
QSP models have considerably evolved and been applied to AD development programs and should be an important component of the MIDD strategy. These tools allow one to pressure test the assumptions behind experimental findings, such as interrogation of the possible non-linear biology for Abeta, the discordant “sweet spot” theory for amyloid decrease, differential impact of Abeta baseline and rate of accumulation on cognitive outcomes (5). QSP models can also identify AD targets with high degree of success, for example, a team of investigators were able to propose the validity of targeting the sphingosine-1-phosphate 5 receptor (S1PR5) as a potential novel treatment option for AD (6).
Nick Holford has advocated disease progression modeling in neuroscience. The key premise is to differentiate between symptomatic and disease modifying drug effects using modeling (7). Current efficacy read-outs for cognitive function assessments include subjective tests. These include the gold standard ADAS-Cog. This battery was originally developed to evaluate cognitive dysfunction dementia. However, within drug development programs, it has been widely used to detect earlier stages of disease progression. Other common batteries include MMSE and CANTAB. While undoubtedly helpful within large well-sized trials, they present inherent challenges when relating to modeling disease progression. Recent use of new methods such as item response theory have helped drive precision around interpretation of ADAS-Cog data (8). This further attests to the significance of a MIDD augmented development program yielding valuable insights to big data.
Given the complexity of AD, a QSP modeling approach is often the most preferred approach. QSP allows us to integrate knowledge and data to quantitatively describe the different biological and physiological mechanisms related to disease progression. As such, this is a very useful tool when trying to understand the effect of drugs with different targets while helping to make decisions on specific points of interventions to optimize possible AD treatments based on mono- or combination therapies (see Figure 1 for a QSP model and the ecosystem of AD opportunities it represents).
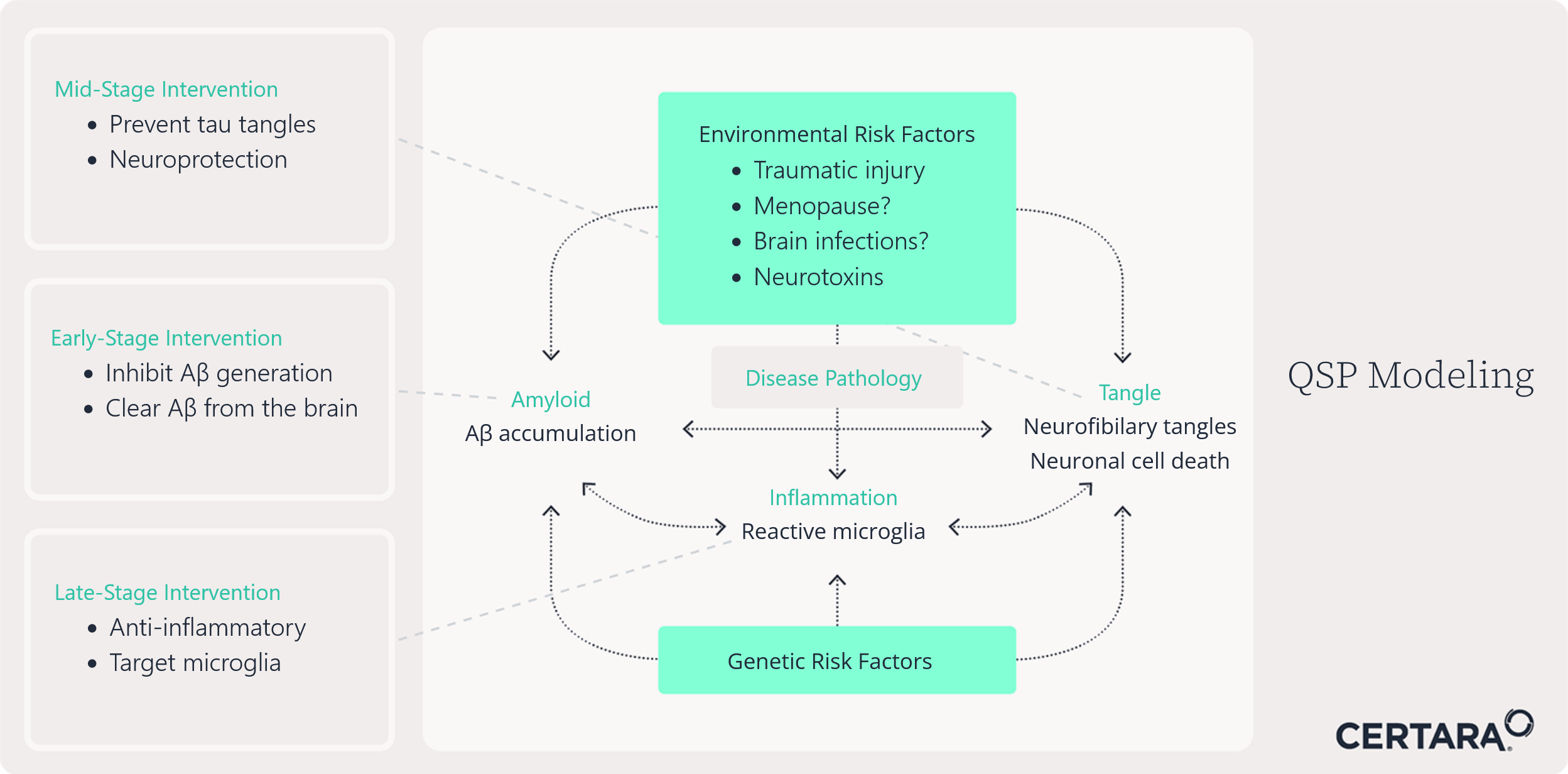
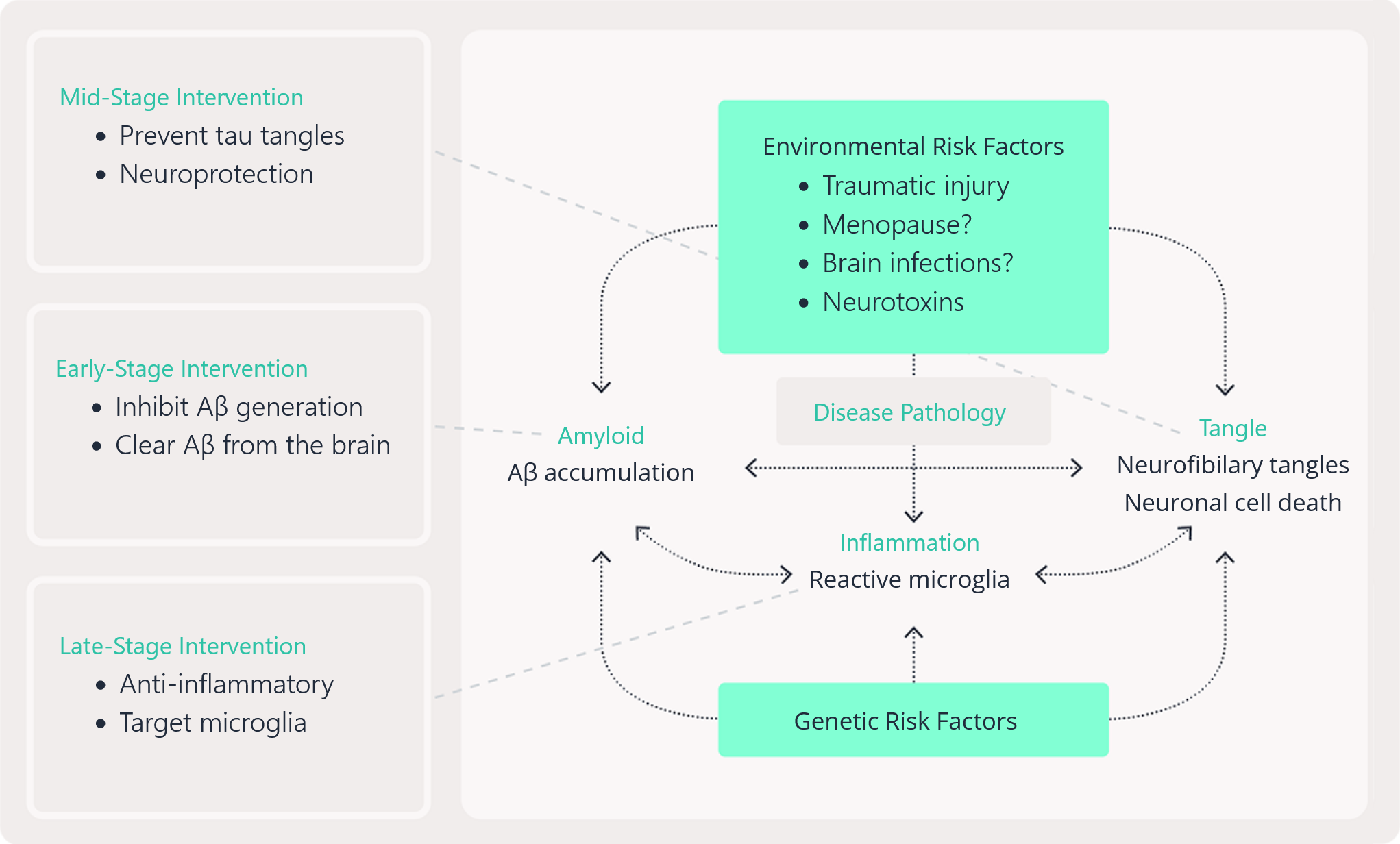
Certara scientists have a wealth of integrated drug development and QSP experience with neuroscience targets, including AD, one of many examples includes a modeling platform which has generated testable hypotheses on the cognitive worsening of BACE inhibitors and the different outcomes for the two Phase III trials with aducanumab in AD (9).
See how Certara IQ™ Alzheimer’s Disease QSP model helps researchers simulate disease progression, therapeutic mechanisms, and biomarker responses.
Advancing Alzheimer’s Disease Clinical Trials and Drug Development Through Non-Animal Methods
One major obstacle has been the reliance on traditional animal models that poorly replicate human Alzheimer’s pathology. As scientific understanding and technological capabilities evolve, non-animal methods are emerging as powerful tools that can revolutionize drug development for Alzheimer’s disease. These human-relevant, ethical, and often more accurate approaches hold immense promise for improving the speed, efficacy, and success rates of therapeutic discovery.
The limitations of animal models, particularly rodents, in Alzheimer’s research are well documented. Mice and rats, which are commonly used in preclinical studies, do not naturally develop Alzheimer’s disease. To mimic the condition, scientists genetically modify them to overexpress human genes associated with amyloid-beta or tau protein production—hallmarks of Alzheimer’s. However, these modifications do not capture the full complexity of the disease as it occurs in humans, especially sporadic late-onset Alzheimer’s, which is influenced by multiple genetic and environmental factors. Consequently, many drugs that show promise in animal models fail in human trials. According to the Alzheimer’s Drug Discovery Foundation, the failure rate for Alzheimer’s drugs in clinical trials exceeds 99%. This alarming statistic underscores the urgent need for more predictive models that closely mimic human physiology and pathology. Non-animal methods, especially those rooted in human biology, offer an innovative path forward.
Human Cell-Based Models and Brain Organoids
One of the most transformative developments in biomedical research has been the advent of induced pluripotent stem cells (iPSCs). These cells, derived from adult human tissues such as skin or blood, can be reprogrammed into any cell type, including neurons. By creating patient-specific iPSC-derived neurons, researchers can study Alzheimer’s in a dish—observing disease progression in real time, under human genetic conditions.
Further enhancing the relevance of these models are 3D brain organoids, miniature, lab-grown clusters of human brain cells that self-organize to resemble aspects of the brain’s architecture and function. Organoids can exhibit key features of Alzheimer’s pathology, such as amyloid plaque accumulation and tau tangles, offering a more comprehensive and physiologically accurate environment for studying disease mechanisms and drug responses. These models not only better mimic the human condition but also enable personalized medicine. Drugs can be tested on organoids derived from specific patients, potentially predicting individual responses before clinical trials. This patient-centric approach could lead to the development of more effective, targeted therapies.
In addition to cell-based techniques, computational models and artificial intelligence (AI) are playing an increasingly critical role in Alzheimer’s drug discovery. AI algorithms can process vast datasets from genomics, proteomics, and clinical trials to identify new drug targets and predict drug interactions. Machine learning can also analyze imaging data to detect early biomarkers of Alzheimer’s, accelerating diagnosis and intervention. Computational modeling of disease pathways allows researchers to simulate how drugs interact with complex biological systems. These in silico models can rapidly screen thousands of compounds, identifying those most likely to be effective and safe—without the need for animal testing. Not only does this save time and money, but it also focuses laboratory efforts on the most promising candidates, improving the likelihood of clinical success.
Organ-on-a-chip technology represents another frontier in non-animal research. These devices use microfluidics to simulate the structure and function of human organs on a chip the size of a USB stick. A “brain-on-a-chip,” for instance, can replicate the blood-brain barrier—crucial for understanding drug delivery to the brain—and the interactions between neurons, glia, and immune cells. This technology allows researchers to observe how drugs affect human brain tissue under controlled, reproducible conditions. Furthermore, multiple organ systems can be connected via microfluidics to study how drugs are metabolized and how they interact across the body. Such systems are far more predictive of human outcomes than animal models, which often differ significantly in metabolism, immune response, and neurophysiology.
Moreover, regulators are increasingly recognizing the value of these approaches. Agencies such as the U.S. FDA and EMA have begun to support the integration of non-animal methods into drug approval pathways, signaling a paradigm shift in biomedical research.
Learn more
Learn more about Certara’s new Non Animal Navigator offering, that can help clients navigate this new development.


Senior Distinguished Scientist, Drug Development Solutions
Rajesh is a scientific key opinion leader with 25+ years in drug development, specializing in model-informed strategies for biologics, vaccines, and small molecules. Currently a Senior Distinguished Scientist at Certara, he leads strategic consulting and the CDDS centers of excellence. Previously, he founded Merck’s quantitative clinical pharmacology department and held key roles at Aventis and Bristol-Myers Squibb. Rajesh holds a PhD in Pharmaceutical Sciences (University of British Columbia) and an MBA in Strategy and Innovation (Warwick). Consistently recognized among the top 2% of influential scientists, his work includes 100+ publications, 89 posters, and 4 books. He is an elected fellow of AAPS.
This blog was originally published in September 2021 and has been updated for accuracy.
Contact us to learn more about the Certara Non-Animal Navigator™ Solution


