



Regulatory submissions must conform to the electronic common technical document (eCTD) format to be successfully received and reviewed by health authorities. This might seem simple. However, a sponsor with insufficient medical writing and regulatory publishing expertise may encounter obstacles and risks with this complex technical process.
eCTD is an interface for industry-to-agency transfer of regulatory information that is used by countries around
the world (Figure 1).
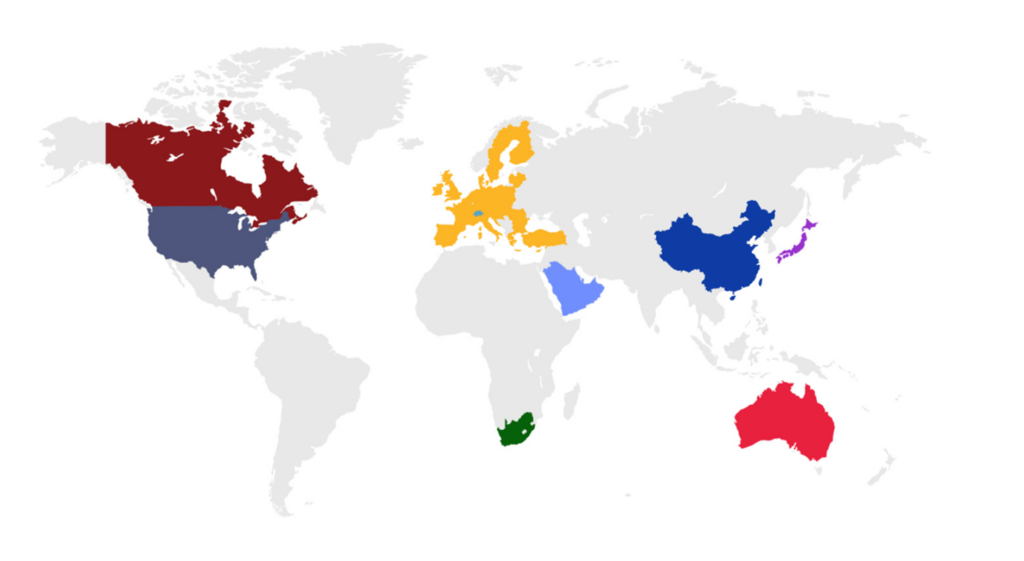
Figure 1. Countries that require submissions in the eCTD format.
This blog post will describe common pitfalls that pharmaceutical sponsors encounter during the regulatory eCTD submission process and some best practices for addressing them.
A Brief History of eCTD
Historically, paper-based regulatory submissions were prepared manually. Over the past decade, the process has evolved tremendously. Technical tools are available to support an entire electronic dossier.
The eCTD submission procedure was developed by the International Conference for Harmonization (ICH) Multidisciplinary Group 2 Expert Working Group, an organization that develops international standards (ISOs).
The standard defines the eCTD submission criteria to be implemented by global health authorities to streamline regulatory review of new drugs, and potentially all regulated products. The eCTD format is organized into five modules:
- Administrative information and prescribing information
- Common technical document summaries
- Quality data
- Nonclinical study reports
- Clinical study reports
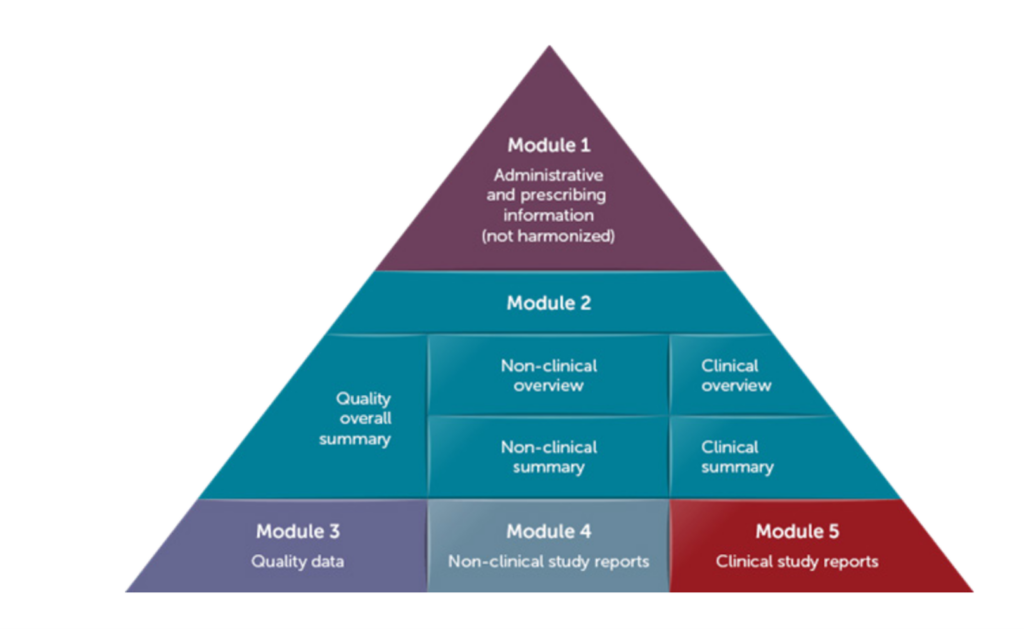
Module 1 Administrative information and prescribing information contains region-specific documents. Modules 2-5 are common to all ICH regions. Module 2 provides an overview and summarizes the data for Modules 3, 4, and 5.
The Sponsor develops the content for modules 2-5 that can be reused for submissions wherever eCTD is accepted: US, Canada, the European Union, etc.
How RTFs Happen & How to Mitigate Your Risk of eCTD Submission Fails
Health authorities issue Refusal to File (RTF) letters to inform sponsors that their application(s) don’t comply with the agency’s requirements. Two common regulatory pitfalls can invoke an RTF.
- eCTD submission fails can result from noncompliance with technical requirements. Upon receipt, the US Food and Drug Administration (FDA) runs submissions through a Global Submit Validate tool. If the submission has serious validation errors (High Errors), they will reject it immediately. An example of a high error is the folder structure, files, or XML not meeting the technical validation criteria.
- Submissions fail based on noncompliance with the content requirements. For example, missing information or summaries, PDFs that don’t follow the required PDF technical standard, or datasets that aren’t in CDISC format.
To be compliant and reduce the risk of an RTF, the application must meet the health authority’s technical and content requirements. Rejected submissions can delay drug approvals and cost sponsors time and money.
Ensuring High-Quality Submissions
Build your eCTD submission with documents that comply with ICH and regional PDF specifications. Having clear expectations and standardized operating procedures to streamline the submission process will eliminate confusion on how to deliver and publish documents.
Creating documents that are eCTD-ready is the first step to ensure a successful submission. Embracing regulatory operations best practices enable sponsors to avoid the risks of failing to meet eCTD standards and thus incurring RTF from an agency.
Incorporating best practices for regulatory writing to support medical writing and project management is essential. When medical writing is complete, documents are prepared for eCTD publishing by our regulatory publishing service team who then manages the submission. The regulatory publishing service team will ensure the submission is valid and will be accepted by the agency.
Satisfying Regional Requirements
eCTD requirements between regions differ in two major ways: differences in module 1 content and the validation criteria.
eCTD module 1 contains regional requirements: prescribing information, packaging, and local government forms (e.g., PDUFA user fees in the U.S.). Each region has submission specifications and requirements. For example, the European Union can accept submissions using a “centralized procedure,” which grants approval to market a New Drug Application in all EU countries.
Additionally, each region has validation criteria for analyzing submissions for errors. Whenever a country or region updates its validation criteria or local requirements, we update the version of our submission validation software for that specific country or region.
Now that you are aware of the complexities involved in developing an eCTD submission, learn more about how to implement eCTD publishing via outsourcing. Please read the white paper that we have written on this topic.
This blog was originally written on July 21, 2017, by Rob Connelly. It was updated by Shawn Harris on March 1, 2024
