



Drug developers often rush through the discovery process to get into human testing. Most are so curious about their new target or molecule that they may forget that the pharmaceutical formulation process that can easily make or break their program. Once in human testing, revisiting these formulations is often time-consuming. By then, depending on drug effects, there may or may not be a desire to “re-invent the wheel.” These formulation challenges then continue and can become unsurmountable to the developer.
Why is drug product development important?
Formulations carry very specific technical and regulatory expectations as they progress from the first formulation to a marketed formulation. For example, you will likely start your first-in-human trial with granules or solution or suspension (some developers refer to this as “drug in bottle”). In phase 2, this formulation would be developed further into a capsule dosage form and then on to a tablet in phase 3. The final market image would likely be an embossed, engraved, and colored tablet dosage form. Each change imparted to the dosage form will change the drug effect and/or safety profile.
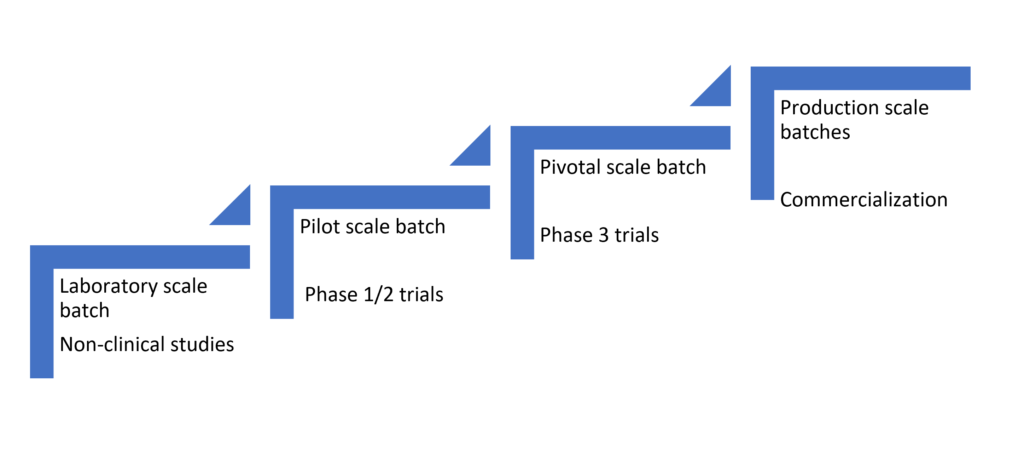
“Batches” denote the type of formulation and often refer to the time period within the drug development journey (Figure 1). For example, a laboratory scale batch is earmarked for pre-formulation studies, drug substance optimization (salt forms, etc.), and is routinely used for non-clinical studies. Pilot scale batches are often used in process development and optimization of drug product, manufacturing process and quality-by-design, and routinely used in early clinical trials (could be drug-in-bottle, suspension, capsule). Pivotal scale batches include scale up and pivotal bioequivalence batch and are routinely used in phase 3 trials – final market composition (no embossing). Production scale batches are those used in the commercialization of final product and are often embossed as the final market image.
You may have noticed that a number is often imprinted on oral dosage forms such as a tablet. Ever wonder what those are? They are “imprint codes.” A pill imprint code is a unique identifier that applies to all solid oral dosage forms such as tablets, capsules, and pills. It is an alphanumeric text which is embossed, debossed, engraved or printed onto solid oral dosage forms. Of course, they may also have other identifying markings, such as trademark letters, marks, symbols, internal and external cut outs. These imprint codes are required by the US FDA!
Also, technical details on formulation type and composition are considered confidential information and proprietary to the sponsor. If you have ever looked through the chemistry, manufacturing, and control (CMC) section of a summary basis of approval document, you’ll notice that this section is the most heavily redacted document of all the documents. See below!

Why do we conduct pre-formulation studies?
Pre-formulation studies typically start when a newly synthesized compound shows clinical promise. These studies examine how the new compound’s physicochemical properties could affect drug performance and help develop a dosage form. A snapshot of these studies and what they address is shown in the following Table.
| Bulk characterization | Solubility analyses | Stability analyses |
|---|---|---|
| • Crystallinity and polymorphism • Hygroscopicity • Fine particle characterization • Bulk density • Power flow properties |
• Ionization constant • pH solubility profile • Common ion effect • Thermal effect • Solubilization • Partition coefficient • Dissolution |
• Stability in toxicology formulations • Solution stability • Solid state stability |
Here are 3 key pharmaceutical product development questions to consider.
#1 Do you have the right salt?
Improving bioavailability is arguably the first question the clinical team asks formulators. Most druggable targets are lipophilic. Hence, molecules with pre-defined pharmacophores are often poorly water soluble and pose challenges to the formulator. Chris Lipinski, a Pfizer thought leader and author of the “Rule of 5” has said: “Poor aqueous solubility is the largest physicochemical problem hindering drug oral activity.” So, what is his rule of 5?
Rule of 5 is “When two or more thresholds of molecular weight (MW) >500, calculated logP >5, a number of hydrogen bond donors >5, or hydrogen bond acceptors >10 were exceeded, oral exposure would be limited by poor solubility and/or poor permeation”.
Preparing salts of an active pharmaceutical ingredient can improve bioavailability. However, identifying the salt form most suitable for further development isn’t easy. It typically involves a salt screening process, limited animal testing, and some biopharmaceutic analyses. To give you a glimpse of the cations and anions drug developers have used, see this Table adapted from Remington’s Pharmaceutical Sciences:

#2 Do you have the right particle size?
Modulating the particle size of the API can fine tune the formulation to optimize drug performance. One may recall the digoxin bioavailability issues in the mid-1970s that was related to change in particle size (Shaw and Carless, 1974).
Reducing particle size is accomplished by milling which is one unit operation within pharmaceutical manufacturing. When a formulation scientist asks a clinician whether to reduce the particle size of the API, the response is usually “we need the most bioavailable form.” This can create significant inefficiencies down the value chain. A more suitable response would be “Can we run an experiment with various particle sizes and see if bioavailability changes without adversely affecting stability?”
It’s not just the mean (or median) particle size that needs to be focused on. The entire particle size distribution needs to be considered because these distributions could overlap and convey an illusion that absorption and hence bioavailability may not differ.
There are variations to the milling process, wet and dry being common. Nanomilling has been a recent manufacturing innovation. The bigger issue is building a specification around particle size – the more variable the effect as a function of PS, the more difficult it would become as the product gets scaled up. Ensuring the optimal particle size is a crucial consideration from both a bioavailability optimization and a scale up point of views.
#3 Do you have the right solid-state form?
Another common way to affect bioavailability improvement is to alter the solid-state form. This is a known scientific discipline of material sciences and is often pursued to assess the most stable form of a pharmaceutical drug substance. One may think this is only to do with the drug substance, but even excipients are affected by solid state polymorphism and in particular, drug-excipient interactions.
Polymorphic forms in their most simplistic sense refers to crystalline and amorphous forms but can include other forms such as solvate and hydrate forms, or co-crystals. Refer to the FDA guidance on this subject.
Now, history is littered with all kinds of issues with polymorphic drug substances. The paper by Newman and Wenslow (2016) highlights the complex interplay between polymorphic states, patent law, and infringement. It’s common to have multiple polymorphic forms during product development. The issue is knowing that they exist and understanding the material effect it may have on product value and performance.
So why is solid state polymorphism important aside from the usual motivator of affecting BA changes? There are many considerations. Polymorphic forms of a drug substance can have different chemical and physical properties. These properties can influence the ability to process and/or manufacture the drug substance and the drug product and may have important effects on issues such as drug product stability, dissolution, and bioavailability. Regulators pay close attention to this issue as it effects drug product quality, safety, and efficacy.
In summary, a plethora of drug substance related characteristics can affect clinical pharmacology properties of drugs. Ensuring that there is adequate understanding of how they could be impacted by formulation switches in development will only prepare the developer to ensure regulatory questions are answered. Many model-informed drug development solutions are available for biopharmaceutics topics such as these. Most notable of these include PBPK modeling using platforms such as the Simcyp Simulator. Certara scientists have experience with such modeling and clinical pharmacology product support. Reach out to us if we can assist in your formulation bridging questions.
References
- Berge SM et al. Pharmaceutical salts. J Pharm Sci. 66(1), 1-16, 1977.
- Gibson M, Pharmaceutical Formulation and Preformulation, Marcel Dekker, 2009
- Newman A, Wenslow R. Solid form changes during drug development: good, bad, and ugly case studies. AAPS Open (2016) 2:2. URL: https://aapsopen.springeropen.com/track/pdf/10.1186/s41120-016-0003-4.pdf (accessed: August 4, 2022).
- Shaw TR, Carless JE. The effect of particle size on the absorption of digoxin. Eur J Clin Pharmacol. 1974 Jul 26;7(4):269-73.
