


The ICH E11A draft Guideline on Paediatric Extrapolation was released on 4 April 2022 entering a public consultation phase. This guideline has several objectives. While aiming to harmonize methodologies and approaches for pediatric extrapolation in drug development, it balances access to pediatric indications and reduction of children’s exposure to experimental medications where warranted. The ICH E11A webpage outlines these considerations.
The extrapolation framework includes an extrapolation concept, followed by a step-by-step extrapolation plan. The latter specifies the relevant studies to be performed as well as analytical frameworks such as model-informed methods as well as use of biomarkers. Ultimately, the guideline addresses the issue of pediatric dose selection.
In this blog, I will reflect on three essential themes that dictate the guideline’s concepts.
Theme #1: Model-informed drug development
The heart of the extrapolation guideline is premised on what model informed drug development (MIDD) has to offer. This is a welcome guideline from that perspective.
There are two preconditions to demonstrate evidence of the similarity threshold: disease similarity and response to treatment similarity.
Disease similarity
In section 3.1., the guidance clarifies that the evaluation of disease similarity does not imply a “sameness” standard but rather “whether the disease is different to a degree that would preclude pediatric extrapolation.” This is an interesting clarification and offers the possibility that the disease could be different and still allow for extrapolation.
Another interesting clarification relates to subgroups and the fact that disease subgroups between adult and pediatrics might be similar when the overall population might not be. The ICH prefers a reasonable dataset for allowing extrapolation vs an airtight case. An example is also shared about similarity in dilated cardiomyopathy as a subset of heart failure.
Several considerations are presented to support the threshold of similarity of disease, but the most meaningful is on the understanding of the natural history of disease and on diagnostic criteria.
Kalaria et al. demonstrate a good example of what constitutes disease similarity (2021).
Figure 1: Comparison of placebo response in adult and pediatric patients. Solid and dashed blue lines represent the pediatric and adult patient populations receiving placebo (Adapted from Kalaria et al., 2021).
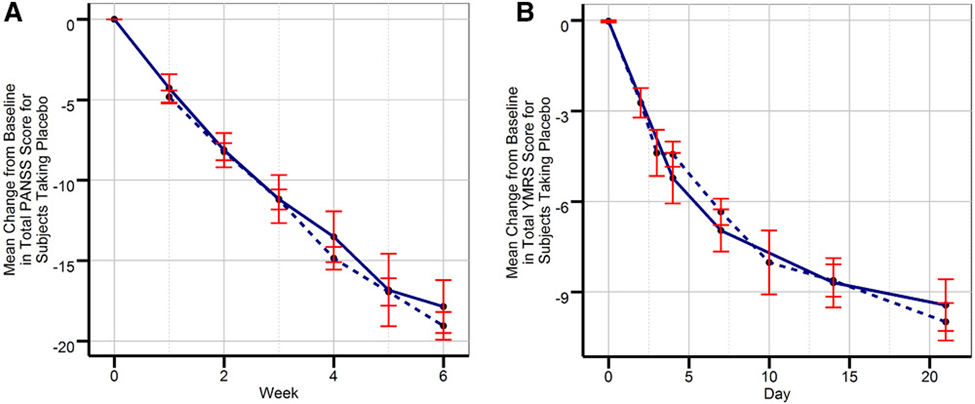
There are two separate but interlinked parts of this standard. One speaks to pharmacology and the other speaks to similarity of response. In the former, the guideline reflects on clinical pharmacology considerations such as how the drug works (mechanism of action) as well as their pharmacokinetic and disposition properties. In the latter, the emphasis is on exposure/response relationships and a greater appreciation of how the drug interacts with a receptor and elicits a pharmacological cascade and how they may be similar or different within the adult and pediatric subsets.
A good example of response similarity is shown in Figure 2 from the work of Mehrotra and coworkers (2016).
Figure 2: Relationship between darunavir inhibitory quotient (IQ) and the probability of virologic success at week 24 in adults and pediatric subjects (Adapted from Mehrotra et al., 2016)
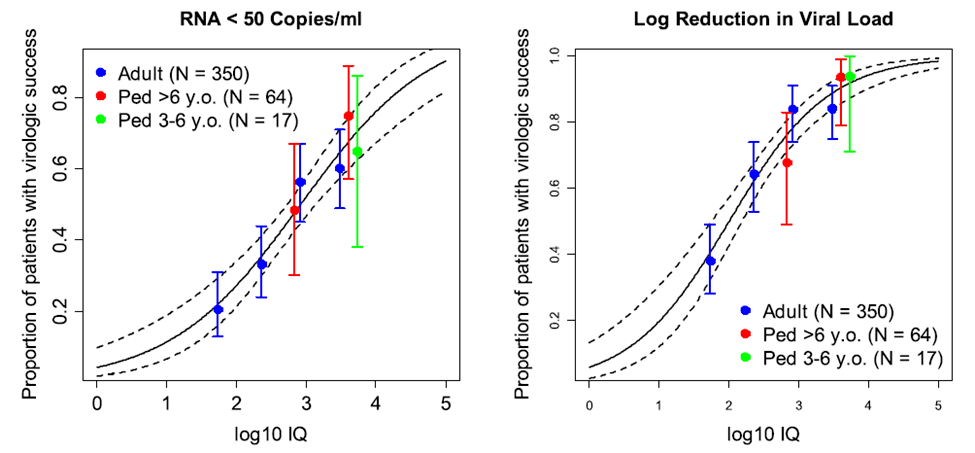
Section 3.4 outlines the various data sources relevant to the use of PK and PK/PD analyses but interestingly includes a real-world evidence (RWE) category. It is unclear whether the agency would expect each of the data sources to be submitted. It is probably unrealistic to expect real world evidence to be part of each submission package, especially for an unprecedented target or a rare disease. The guideline does caveat RWE to be an “evolving” consideration.
To execute on this overarching strategy, the extrapolation plan considers three aspects, 1) adequate and well controlled trials, 2) use of biomarkers, Bayesian vs frequentist approaches and PK/PD, and 3) exposure matching considerations. Section 4 explains the extrapolation plan in detail.
Dose selection is prominently featured in this section, with particular emphasis on the physiologic maturation of targets and organ systems that may affect the way a drug action is characterized. The use of biomarkers is encouraged, and the guideline is open to both validated and unvalidated biomarkers. The use of mechanistic modeling and quantitative systems pharmacology (QSP) analyses are welcomed. Two scenarios (section 4.1.3.) are presented for dose selection for establishing efficacy, one based on PK alone and the other based on biomarkers alone. In the first, exposure matching appears sufficient (with clear knowledge of acceptable target exposure metrics), and in the second, biomarkers can be used in lieu of exposure matching. One precondition to using the biomarker approach is a relationship between the biomarker and the efficacy endpoint.
Exposure matching
While the guideline does not provide details on exposure matching, the paper by Mulugeta and coworkers is instructive (2017). This seminal and comprehensive work evaluated approaches used for matching adult systemic exposures as the basis for dose selection in pediatric trials (FDA). Figure 3 is abstracted from this work and shows how the PK parameter ratios, Cmax and AUC, compare for pediatric/adult data.
Figure 3: Pharmacokinetic parameter ratios for products approved at the same dose (adapted from Mulugeta et al., 2017)
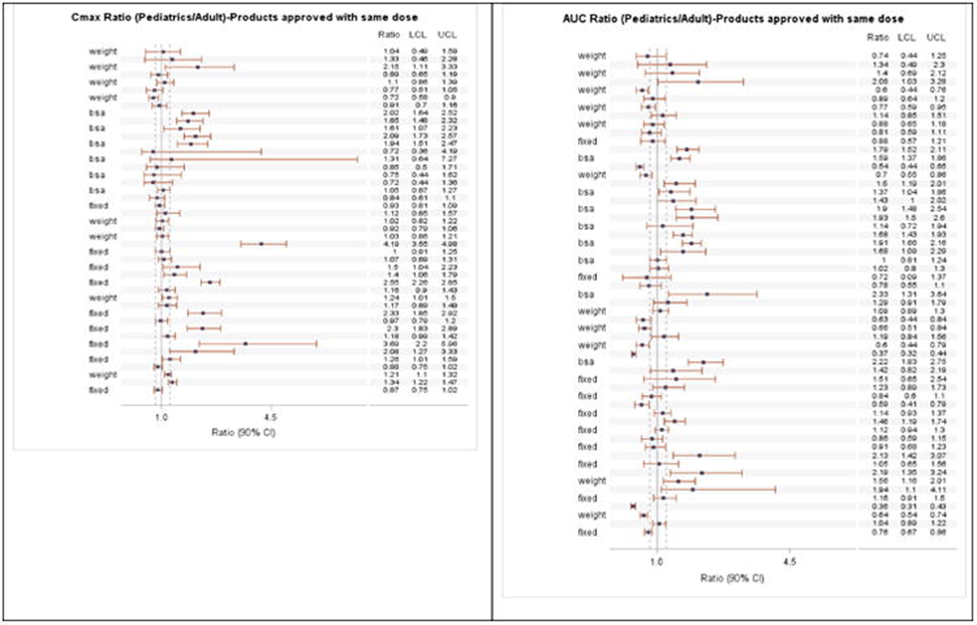
While the pediatric extrapolation guideline mentions that adult to pediatrics similarity should be assessed, it stops short of specifying acceptable methods. This approach means that any sponsor can present their rationale for the similarity method used.
In principle, however, there are only two possible approaches to assess similarity regarding exposure matching. These include the conservative standard, which is the bioequivalence standard where the no effect boundary of 90% CI is applied to the 80-125% bounds for AUC and Cmax. Alternatively, a model-informed approach can be utilized using a dose/response or exposure/response in that Emax, EC50 and/or the slope can be within acceptable limits (e.g., numeric bounds or distribution of the pediatric data within the overall adult data).
As mentioned earlier, model-informed drug development features prominently in this guideline and section 4.2 details the types of tools that can be applied to the extrapolation plans. Section 4.3 briefly reviews the types of efficacy studies that are to be designed to obtain efficacy in a pediatric extrapolation plan. An interesting subtext is in section 4.3.7. which differentiates between frequentist and Bayesian designs.
Theme #2: Inclusion of adolescents in adult pivotal trials
Probably the most important aspect to the guideline is in Section 5.2, which recommends including adolescents in adult clinical trials. This is important to pediatric drug development because it will accelerate the access to important medicines to pediatric populations. Traditionally, the drug development engine is built around incremental risk, and the most conservative enterprises tend to study populations in a linear fashion, i.e., study adults completely before launching a pediatric investigation. Inclusion of adolescents 12 to 17 years of age in adult trials can greatly increase the confidence in the pediatric/adult pivotal dataset analyses for efficacy, however more importantly, for safety. The guideline asks for pharmacological basis to be addressed, in that both the similarity of disease as well as response to treatment are both similar between adult and adolescents. When the natural history of disease is not well known, this aspect can be best determined by the adolescent cohort being incorporated much earlier than the pivotal studies. It will be interesting to see how the sponsors react to this part of the guideline and include adolescents as part of their adult late stage development. One consideration is to include pediatric subjects down to 5 years of age if the ontogeny of drug disposition is not expected to be meaningfully different from adult life.
Theme #3: Extrapolation of safety
The guideline speaks to safety in sections 3.5 and 5.1. Prior to this guideline, extrapolation was generally accepted to be applied to conditions of disease similarity and response similarity. However, understanding how safety can be extrapolated to the pediatric population required assessing whether there are compound vs mechanism-based safety considerations, and whether there are unique considerations in the pediatric population that is more related to developmental ontogeny, i.e., that pediatric population might in fact be more sensitive than the adults. This guideline provides for the possibility of extrapolation of safety which makes it a rather provocative and interesting consideration.
In summary, while the extrapolation framework is discussed in the guideline, it lacks the specificity that sponsors of pediatric investigational plans will expect.
For more information on pediatric extrapolation, read the blog I authored previously.
Blog
References
Kalaria SN, et al. Extrapolation of Efficacy and Dose Selection in Pediatrics: A Case Example of Atypical Antipsychotics in Adolescents With Schizophrenia and Bipolar I Disorder J Clin Pharmacol. 2021 Jun;61 Suppl 1:S117-S124. doi: 10.1002/jcph.1836.
Mehrotra N et al., Role of Quantitative Clinical Pharmacology in Pediatric Approval and Labeling. Drug Metab Dispos 44:924–933, July 2016.
Mulugeta Y, Barrett JS, et al. Exposure Matching for Extrapolation of Efficacy in Pediatric Drug Development. J Clin Pharmacol. 2016 Nov;56(11):1326-1334. doi: 10.1002/jcph.744.
