



This is the second of three blog posts discussing best practices for developing oligonucleotide therapeutics. The first blog in the series explained how these therapies work.
Characterizing the pharmacokinetics (PK) of an investigational oligonucleotide drug is an important part of its pre-clinical development program as well as informing its later clinical development. In this blog, I’ll explain several common pre-clinical challenges in describing the ADME (absorption, distribution, metabolism, and excretion) of this therapeutic modality and how modeling and simulation approaches (M&S) can help address them.
Pharmacokinetic (PK) Considerations
Oligonucleotide PK characteristics are governed by three factors: their type of modification, their conjugates, and for small interfering RNAs (siRNAs), their carriers.
For example, antisense oligonucleotides (ASOs) that contain a phosphorothioate backbone are extensively bound (more than 85%) to plasma protein, which is predominantly albumin. That is true across most species, including humans. That high degree of binding reduces urinary excretion and renal clearance of the drug because it does not go through glomerular filtration. However, the binding affinity to albumin is relatively low, which allows uptake to the tissue.
siRNA drugs are less charged and less bound to plasma proteins, so they are cleared rapidly from the blood either by nuclease metabolism or excretion in urine. For example, less than 2.1% of patisiran is bound to plasma proteins, and the terminal elimination half-life for this drug is only three days.
With respect to conjugates, if you compare antibody-drug conjugates (ADCs) with GalNAc-siRNA conjugates, such as fitusiran and inclisiran, those single-chemical entities are targeted to the liver and allow for easier subcutaneous (SC) administration.
The carrier also has an impact. The lipid nanoparticle (LNP) delivery system for patisiran, for example, is approximately 85 nanomolar, allowing targeted delivery in the liver after intravenous (IV) administration.
Using Preclinical Information to Inform Clinical PK
Absorption
After IV or SC injection, single-stranded ASOs transfer from blood into tissue in minutes to hours. Their rapid transmission into the cell is predominately facilitated by endocytotic uptake. Once intracellular, ASOs exhibit long half-lives (two to four weeks) and prolonged activity in suppressing or altering expression of the target RNA.
Following SC administration, ASOs are absorbed from the injection site into the circulation, and peak plasma concentrations are consistently reached within three to four hours. Nearly complete bioavailability has been observed with multiple ASOs after SC administration, at least in monkeys.
Then, plasma concentrations decline from peak concentrations in typical multi-exponential fashion. That is characterized by a dominant initial rapid distribution phase, where the drug transfers from the circulation into the tissues in minutes or hours, followed by a very much slower terminal elimination phase where the half-life can be up to several weeks.
The apparent terminal elimination rate observed in plasma is consistent with the slow elimination of ASOs from tissue. This indicates that equilibrium has been reached between the post-distribution phase plasma concentration and tissue concentrations.
But analyzing that data is not always easy. These graphs illustrate one of the challenges that can occur when studying oligonucleotide absorption.
Issue: Absence of a Post-distribution Phase
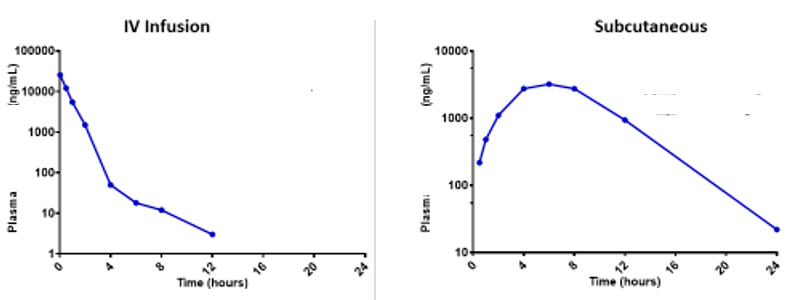
What happened? It is possible that PK sampling did not continue for long enough. The post-distribution phase should be 100+ hours. If the anticipated multi-exponential decline is not captured after SC administration, it could lead to accidental scaling of the flip-flop kinetics and extrapolating the absorption rate rather than the elimination half-life to humans.
Solution: Use modeling and simulation (M&S) to describe the plasma IV and SC administration simultaneously and leverage the strengths of both datasets.
Be aware of possible terminal phase bias. It can appear faster than it is. To model that correctly, the absorption component (bioavailability) needs to be decoupled from the clearance component. It is the clearance component that is going to scale directly to body weight across preclinical species.
In this case, the pre-systemic, mono-exponential absorption phase after SC injection could inform the initial pre-systemic disposition. Then, the slower half-life or two-phase distribution from the IV data could inform the systemic elimination of the drug.
Distribution
The partition ratios between liver and post-distribution plasma concentrations are about 5,000:1 for many modified ASOs across most animal species. Therefore, post-distribution plasma concentrations can provide a surrogate for tissue exposure in humans.
For all animal species evaluated, ASOs distribute broadly into most tissues, except for the central nervous system (CNS), after systemic administration. The major tissues for systemic distribution include the liver, kidneys, bone marrow, adipocytes (the cell body–but not the lipid fraction) –and the lymph nodes.
Issue: Post-distribution Phase Profile is Different in the Plasma and Tissue
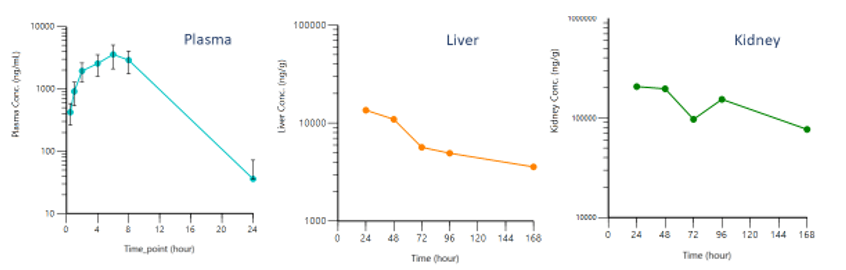
This is an example of a sampling time or below the quantifiable limit (BQL) effect. After 24 hours, the drug level in the plasma sample drops BQL so no long post-distribution equilibrium phase appears in the data. Taking the 10,000 value and dividing it by 5,000, the plasma partition ratio, produces about 2 ng/mL of drug in the plasma, which is below the detection limit. Yet, liver and kidney analyses produce results that are consistent with those in the literature, and a long half-life that is similar across tissues. It is certainly much longer than the half-life seen in the plasma.
Solution: Use M&S to describe the plasma and tissue simultaneously, leveraging the strength of each dataset to reconcile these kinetic profiles.
Pre-systemic data can inform the absorption rate and bioavailability of the drug, while IV data inform the initial, rapid, systemic distribution. Then, tissue data can describe the slow, long terminal half-life, even across plasma where they could appear BQL.
Metabolism and Elimination
Plasma post-distribution PK scales approximately by body weight from monkeys to humans. Tissue concentration between monkeys and humans is also similar on a mg/kg basis due to the linear proportional relationship between body weight and clearance. This holds true across many preclinical species, except mice.
Rodents, in general, are also more sensitive to class-related pro-inflammatory effects of ASOs, than primates, including humans. Therefore, for both clearance and sensitivity reasons, rodents are not considered appropriate species for calculating systemic safety margins for humans.
In vivo experiments in mice have demonstrated that slowly infused ASO drugs produce a substantially greater uptake in the liver than bolus administrations. However, there was little ASO activity associated with those higher concentrations in the liver, suggesting that a nonproductive update pathway exists in cells that endocytosed ASOs presented at low concentrations.
Better in vivo uptake in the liver was facilitated by bolus injections of the ASO because they could overcome the saturable nonproductive pathway.
Using nonsense ASOs to compete with this nonproductive pathway produced less bulk uptake in the liver, and paradoxically, greater antisense activity, as measured by the target RNA silencing, despite the lower total exposure. This suggests at least two uptake pathways in cells, in vitro and in vivo, one leading to productive antisense activity and the other resulting in nonproductive uptake of the ASOs.
Early endosome uptake and sorting pathways may explain this. Further, intracellular binding may be associated with some localization or trafficking of the ASO. The ASO uptake in these cells implicates endocytosis as a dominant update mechanism requiring binding to surface proteins.
DNA also plays an important role in the productive uptake of oligonucleotides as both the base and deoxy sugar are required for optimal update. Nanomolar concentrations of ASO taken up by endocytosis reduce target RNAs within hours, and maximum knockdown occurs within 24 hours, which resemble in vivo observations.
Issue: Assuming the siRNA Half-life and Effect (PD) are Directly Related
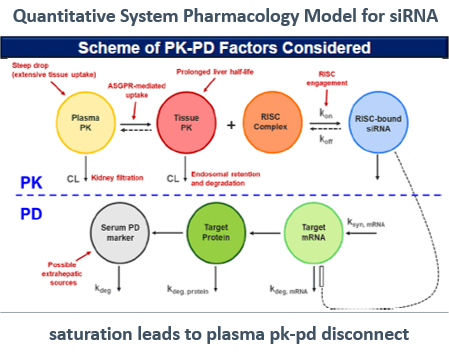
The duration of PK sampling for an siRNA can be shorter than for an ASO, but the duration of PD sampling should be longer. That is because the total elimination half-life of the siRNA is shorter than the half-life of the ASO, due to ASO distribution in tissue and its slower clearance from tissues.
Often though, siRNA therapeutics have a short elimination half-life (up to five days) but the dosing frequency intervals can be up to six months. Whereas the dosing frequency is weekly or monthly for ASO drugs. Even though the clearance is extremely fast, the dosing can occur less frequently due to the long terminal half-life and tissue accumulation.
There is a temporal disconnect between the plasma PK of oligonucleotides and their prolonged PD effect. Choosing the dosing interval and making dosing adjustments are mainly guided by the length of drug effect and PD covariates. Do not use the duration of drug exposure. For example, although inclisiran is eliminated in about 48 hours, a six-month dosing interval was used in the Phase III study and on the label.
Solution: Use M&S to describe the PK and PD simultaneously by adding PD and tissue data to the IV and SC data in the model.
The exact framework and methodology used will depend on the data available and the clinical objective.
Conclusion
M&S offers a practical solution for better understanding these interrelationships, ensuring that preclinical data are accurately interpreted and appropriately applied to advance oligonucleotide drug development.
To learn more about best practices in oligonucleotide drug development, please watch this panel discussion between experts in the field.