



Human mass-balance studies are also called “C-14 studies” or “Absorption, Metabolism, and Excretion (AME) studies.” Drug developers conduct these studies of radiolabeled drugs to quantify the routes of elimination of the drug substance and characterize its metabolism.
The FDA released its first guidance on human mass-balance studies recently. Both Sponsors and Regulators gain extensive insight from human AME (hAME) studies into the disposition of investigational agents before undertaking pivotal clinical studies. Figure 1 illustrates the type of information a typical human mass-balance study generates (Spracklin et al., 2020).
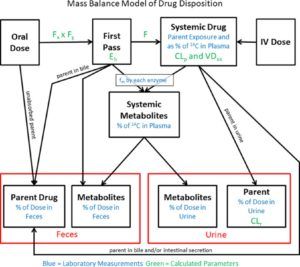
Understanding the hAME properties of a drug is necessary to further inform key clinical pharmacology aspects in a drug development program. These include, but are not limited to:
- an understanding of the plasma drug metabolites forming a bridge to the metabolite exposure of the preclinical toxicology species
- identifying the main elimination pathways based on parent drug and metabolites excreted in the urine or the feces,
- providing information on the minimum absorbed fraction, potential for biliary excretion being a major pathway, etc. (Figure 1).
Knowing the elimination pathways then informs the need for drug-drug interaction (DDI) studies, in particular if mapping enzymes and transporters involved in vitro in the observed major pathways, and whether organ impairment may drastically affect the pharmacokinetics of the drug. Knowledge of the hAME profile can thus assist in setting up the inclusion and exclusion criteria in pivotal clinical trial protocols.
Usually, drug recoveries are >90%. When that happens, no further characterization is necessary. When the drug recoveries are lower than this threshold, and no strong justification for this can be outlined, further studies may be needed to understand the routes of elimination, e.g., when there is a volatile metabolite that is exhaled.
The FDA’s issuance of this document will help harmonize the methods as well as place the hAME study as central to characterize the disposition of a new molecular entity (NME). In exceptional cases, such studies may not be feasible or necessary, and such aspects are handled on a case-by-case basis.
In this blog, we will review and reflect on the scientific and strategic parts of the FDA guidance. We will also reflect on this guidance from a European Union (EU) perspective.
FDA guidance on mass-balance studies
Section III of the FDA’s new draft guidance, lines 60 and onward, states that mass-balance studies should be performed for all molecular entities. However, if a study isn’t done, then provide adequate justification. This is interesting as such studies are not considered to be mandatory. This section also provides avenues where such exceptions can be availed. Lines 69-70 indicate that literature and/or existing product labeling could help a sponsor to argue that a new AME study might not be required. Typically for a sponsor to use such information, they must obtain a right of reference from the innovator IND owner. This could be relevant for marketing applications of active metabolites, enantiomers, etc.
It is conceivable that antibody drug conjugates with identical cytotoxic payloads could use the approved labeling text on hAME based on the letter of the guidance.
Lines 72-74 suggest that biologics, endogenous substances, and peptides may be exempt from hAME studies.
Lines 76-79 suggest that NMEs with >90% recovery in urine of unchanged parent drug could receive study waivers as are those with documented no or negligible systemic exposures. No thresholds of what may constitute as negligible are provided.
Alternatives to hAME studies are also provided in this section.
Timing of mass-balance studies
The guidance emphasizes that mass-balance studies should be done early in clinical development. These studies should be performed prior to initiating late phase clinical trials. Drugs for accelerated approval pathways and/or those that are developed in oncology may have a shorter runway as compared to non-oncology indications. While such a timing may be feasible in the former situations, in the latter, it is not known where the chosen drug candidate is druggable, i.e., full appreciation on whether it has the necessary safety and efficacy properties may not be available. Some sponsors may elect to wait until such information is available, for example, end of Phase 2b dose ranging finding studies to decide on executing such a study.
hAME study design best practices
There is generally no controversy here. Typically, hAME studies that have been conducted prior to this guidance being available were open-label, single dose studies in a handful of healthy volunteers (N~6). That is done to limit subjects’ radiation exposure while gaining an understanding of how the drug is disposed in the human body. The safe absorbed dose of radioactivity is estimated by dosimetry calculations from animal studies, and the dose of the investigational drug should be within the linear portion of the clinically relevant dose range. Often the exact Phase 3 dose may not be available in time for the start of such studies.
The FDA guidance calls for multiple dose studies when time-dependent pharmacokinetics are observed. This conservative approach is aligned with EU requirements and may be relevant if the time-dependency is substantial.
Bioanalytical Assessments
For quantitative profiling of radioactivity in plasma, urine, and feces, samples should be analyzed separately for each subject, which is aligned with the industry best practices. The study report should present plasma and whole blood concentration versus time profiles of total radioactivity, which will be utilized to derive the appropriate pharmacokinetic parameters. The cumulative percentages of the administered radioactive dose recovered in urine, feces, and total excreta should also be presented as a function of time.
In addition to unchanged parent drug measurements, metabolite profiling should be performed in plasma and excreta, which is usually done after pooling the samples in the matrix of interest across time points within each subject. Although, in certain cases, pooling samples across subjects could be the preferred option (e.g., low level of metabolites).
Ideally, at least 80% of the total radioactivity recovered in the excreta should be identified, and, outside of oncology indications under ICH S9 guidance, metabolites accounting for more than 10% of the total drug-related exposure in plasma should be structurally characterized to fulfill the MIST (Safety Testing of Drug Metabolites) guidance requirements. In addition to the percent of unchanged parent and metabolites associated to each matrix, provide a biotransformation scheme with the structures (or tentative structures) of the prominent metabolites.
An EU perspective on mass-balance studies
The EU requirements for mass-balance study design and its interpretation are outlined in the EMA Drug interaction Guideline (CPMP/EWP/560/95/Rev. 1 Corr. 2**). A human mass-balance study provides quantitative information on the major elimination pathways through metabolism, biliary and renal excretion, as well as key metabolite exposure data. This information bridges to the preclinical documentation. In absence of a mass-balance study, required information usually gained in this study needs to be obtained using other sources. If information on metabolite exposure is needed to bridge to species used in preclinical tox studies, then this is usually challenging. The benefits of performing mass-balance studies for oncology drugs to characterize the drug elimination informing potential drug-drug interactions and special populations recommendations are highlighted in the EU oncology guideline (EMA/CHMP/205/95 Rev.6).
An overview of the EU view and reviews of mass-balance studies is well described in the paper by Coppola et al 2019. The draft FDA mass-balance guideline is generally well aligned with the EU view. For drugs with marked nonlinear PK, the EU guideline raises the need to perform the mass-balance study at doses giving rise to the same level of saturation/inhibition/induction as during clinical use to obtain relevant pathway contributions and metabolite exposures. Furthermore, information on excretion of unchanged drug in feces is to a large extent used as information on potential biliary excretion. Two parallel worst case scenarios are evaluated in this respect – full amount of drug in feces residing from either the unabsorbed drug, or from biliary excreted drug. If the fraction of drug in feces is high, these scenarios give rise to very different outcomes and requirements for further studies. In essence, overall, as to be expected, the requirements and regulatory view of mass-balance studies are well harmonized.
Do you need support designing and interpreting the results of a mass-balance study? Our clinical pharmacologists can help!
References
- Coppola P, Andersson A, Cole S. The Importance of the Human Mass-balance Study in Regulatory Submissions. CPT Pharmacometrics Syst. Pharmacol. (2019) 8, 792–804.
- CPMP/EWP/560/95/Rev. 1 Corr. 2** European Medicines Agency. Drug interaction guideline on the investigation of drug interactions.
- EMA/CHMP/205/95 Rev. 6. European Medicines Agency. Guideline on the evaluation of anticancer medicinal products in man.
- Spracklin DK , Chen D , Bergman AJ , Callegari E, Obach RS. Mini-Review: Comprehensive Drug Disposition Knowledge Generated in the Modern Human Radiolabeled ADME Study. CPT Pharmacometrics Syst. Pharmacol. (2020) 9, 428–434.

