


September 15, 2025
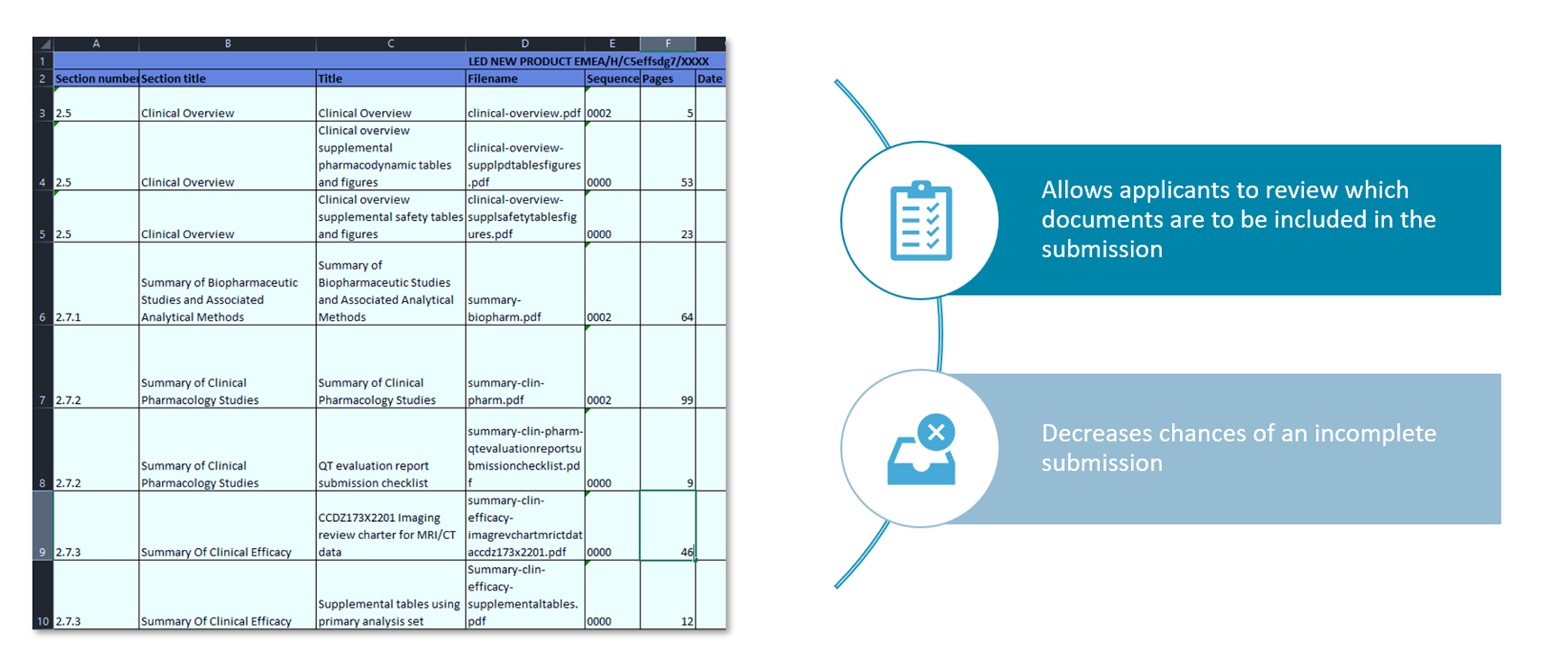
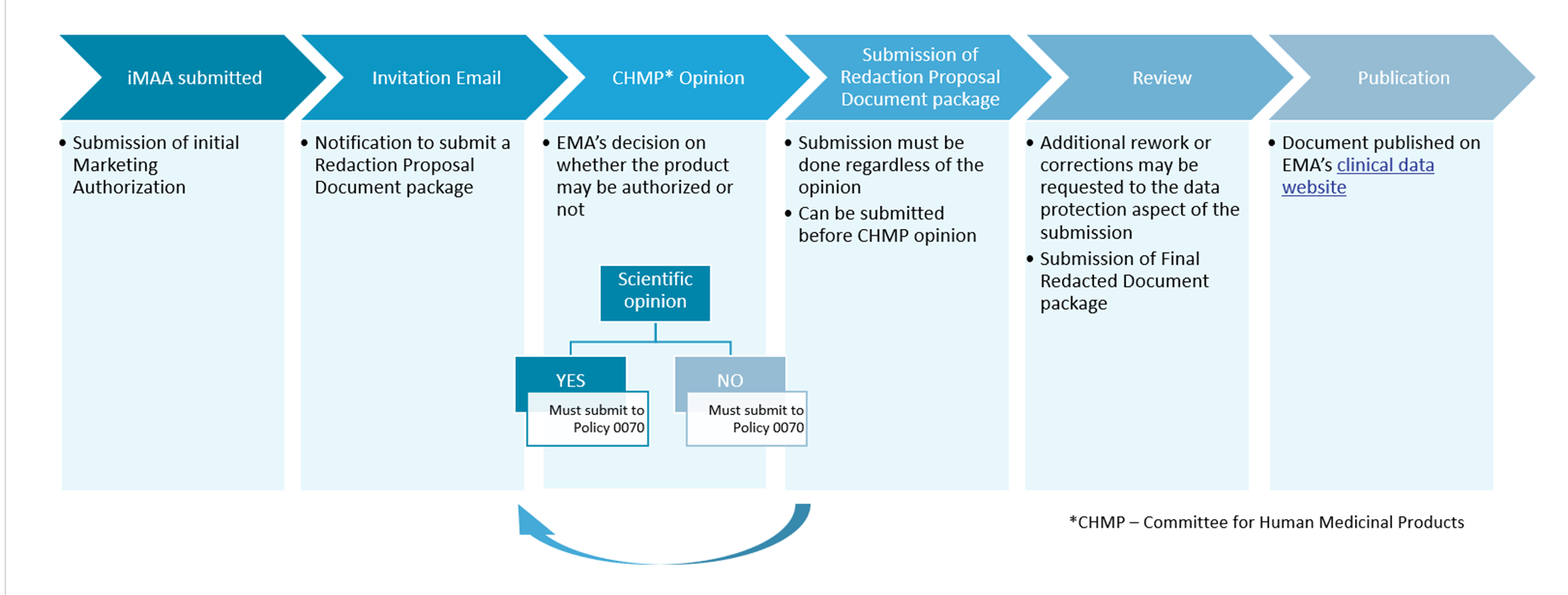

Clinical Trial Data Sharing Under EMA Policy 0070
Read our free white paper and learn how to save time and resources while publicly disclosing clinical trial data.

Senior Transparency Specialist
With a research background in neuroscience, and experience in health information technology, Honz Slipka has a thorough understanding of data analysis, regulatory standards, and best practices in the field of clinical data privacy. Drawing from his experiences, Honz is a champion of innovation, helping to lead the field of science, healthcare, and research into the modern age of technological efficiency, clinical transparency, and data utility.

Document Manager
Bramwell Adams is a Document Manager at Certara. He has extensive experience with EMA Policy 0070 submissions which began from the beginning of Policy 0070 and now has led a good number of successful submissions. Previously, he has worked in the pharmaceutical industry for some 20 years in various document publishing roles.
This blog was originally published in December 2023 and has been updated for accuracy and comprehensiveness.


