


July 1, 2025
The Increasing Popularity of the RSABE Approach
Reference-scaled average bioequivalence (RSABE) is a statistical approach increasingly adopted to assess bioequivalence for highly variable drugs (HVDs). These are drug products for which the within-subject (intra-subject) variability in pharmacokinetic measures—AUC and/or Cmax—exceeds 30% coefficient of variation (C.V.).
In simpler terms, when the same individual takes the same drug under nearly identical conditions (e.g., same dose, administration route, fasting state, and time of day), the AUC and Cmax values are generally expected to be consistent. However, for highly variable drugs, the rate and extent of absorption can fluctuate by more than 30% between administrations—even under controlled conditions—making it challenging to demonstrate bioequivalence using traditional average bioequivalence (ABE) methods.
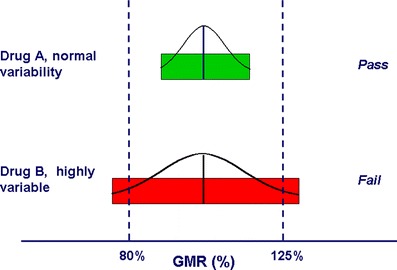
Figure 1, The 80–125% BE limits are represented along the x-axis as two “goal posts.” The BE limits are compared to the hypothetical 90% confidence intervals of the test/reference BE measure GMRs for a drug with normal variability (Drug A) and an HV drug (Drug B). Highly variable drug B would likely have met the BE limits if more subjects had been used. Source: Davit, B.M., Chen, ML., Conner, D.P. et al. Implementation of a Reference-Scaled Average Bioequivalence Approach for Highly Variable Generic Drug Productvaribale graphs by the US Food and Drug Administration. AAPS J 14, 915–924 (2012). https://doi.org/10.1208/s12248-012-9406-x
For highly variable drugs, applying the traditional average bioequivalence (ABE) approach with a standard sample size often fails to demonstrate bioequivalence—even when the test and reference products are genuinely comparable (Drug B in Figure 1). In fact, some HVDs have failed to show bioequivalence even to themselves under standard ABE designs.
This is because high intra-subject variability can obscure real similarities between products, necessitating very large sample sizes to achieve statistical power. Such studies are not only more expensive and time-consuming but also expose more participants to unnecessary risks. As a result, the development and approval of generic versions of HVDs can be significantly delayed or even abandoned, reducing access to affordable alternatives.
The RSABE approach addresses this challenge by scaling the bioequivalence limits according to the within-subject variability of the reference drug. In essence, the permitted acceptance range widens as variability increases, reflecting the inherent pharmacokinetic variability of the drug rather than penalizing it. RSABE can be applied when the within-subject C.V. for the reference product is at least 30%, allowing for a more practical and scientifically justified path to establishing bioequivalence for HVDs.

Want to deepen your understanding of AUC and other PK/PD concepts? Get free access to Pharmacokinetic and Pharmacodynamic Data Analysis ebook.
Why the Method is Called “Scaled”: A Look at the Equations
To understand why the RSABE methodology is referred to as “scaled,” it helps to review the equations behind both conventional and scaled bioequivalence assessments.
Conventional ABE Approach
In Average Bioequivalence (ABE), a drug is considered bioequivalent if the 90% confidence interval for the difference between the logarithmic means of the test and reference products lies within preset regulatory limits (80%–125%). On the log scale, this criterion is expressed as:
OR
These fixed boundaries correspond to approximately ±0.223 on the natural log scale.
Reference-Scaled ABE (RSABE)
For highly variable drugs (HVDs), where within-subject variability (CVWR) is ≥30%, RSABE allows the acceptance range to be widened proportionally to the variability of the reference product. Both the difference and the limits are scaled. The limits are scaled by a regulatory constant, and the difference is scaled by the within-subject variability for the reference product.
EMA RSABE Methodology
EMA RSABE methodology [1] scales the acceptance range by the regulatory constant of 0.294 (which is the SWR at 30% CV) and the difference is scaled by the drug’s variability SWR
which simplifies to:
This means the permissible difference increases with variability, up to a maximum of 69.84%–143.19%.
FDA RSABE Methodology
The FDA methodology [2] applies the same conceptual scaling but uses a different regulatory constant: 0.25. In addition, the FDA uses a more complex statistical test to conclude bioequivalence (not detailed here), but the underlying scaling logic is
which simplifies to:
Therefore, the FDA allows the acceptance range to widen even further than EMA
Reference-scaled Average Bioequivalence acceptance criteria Ranges at Different Variability Levels
Using the formula:
we can calculate the resulting acceptance limits determined by
respectively for EMA and FDA, at various levels of within-subject variability:
Want to deepen your understanding of AUC and other PK/PD concepts? Get free access to the 5th edition of Prof. Johan Gabrielsson’s renowned reference, Pharmacokinetic and Pharmacodynamic Data Analysis.

Executive Director, Training & Certara University
Ana leads the Certara University team in providing modeling and simulation for new drug development through education, skills, and expertise in the global healthcare industry. Ana has more than 20 years experience in a variety of roles in the industry. She has extensive experience in pharmaceutical training, software demonstration, software support, and product management, Ana is also an adjunct faculty member at Skaggs College of Pharmacy and Pharmaceutical Sciences at the University of Colorado.
References
[1] EMA guideline: Product-specific bioequivalence guidance
[2] FDA Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA Guidance for Industry
This blog post was originally published in May 2016 and has been updated for accuracy and comprehensiveness.
Schedule a demo of Phoenix® PK/PD Platform


