



New biologic development and approval of novel biological medications have significantly moved the needle for addressing unmet diseases. Scores of biologics scientists and developers have supported this modern-day marvel.
However, a drug is only helpful if patients can access it. For example, monoclonal antibodies cost thousands of dollars per year. For rare, life-threatening diseases, these medicines may cost up to USD 400,000 per year making them out of reach for large swathes of society.
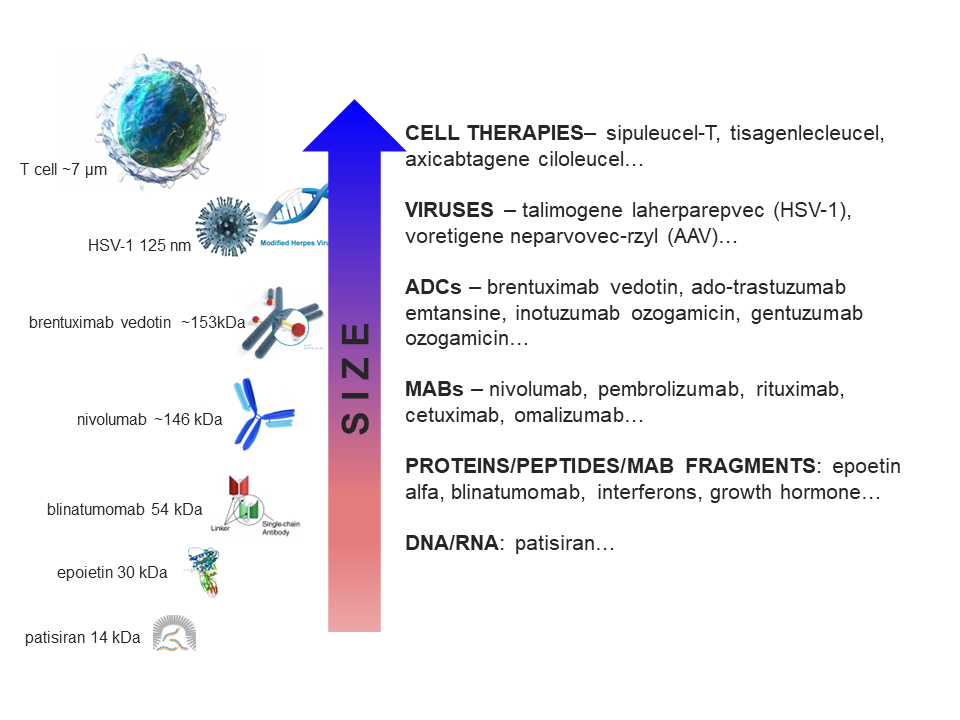
Figure 1. Biologic drugs are larger than small molecule drugs and come in all shapes and sizes.
Biosimilar products have the same efficacy as the innovator but at a much lower price. Thus, the US government enacted the 2009 Biologics Price Competition and Innovation Act to facilitate patient access to lower-cost medicines.
What’s a biosimilar drug?
A biosimilar is a biological product similar to an existing regulatory health agency‐approved reference product.
In Europe, the biosimilar pathway refers to a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product, where similarity to the reference product has been established based on comprehensive comparability.
In the US, the 351(k) Biologics License Application is an abbreviated approval pathway for licensing biosimilar and interchangeable biological products. Section 351(i)(2) of the Public Health Service Act defines biosimilarity to mean that the biological and reference products are highly similar.
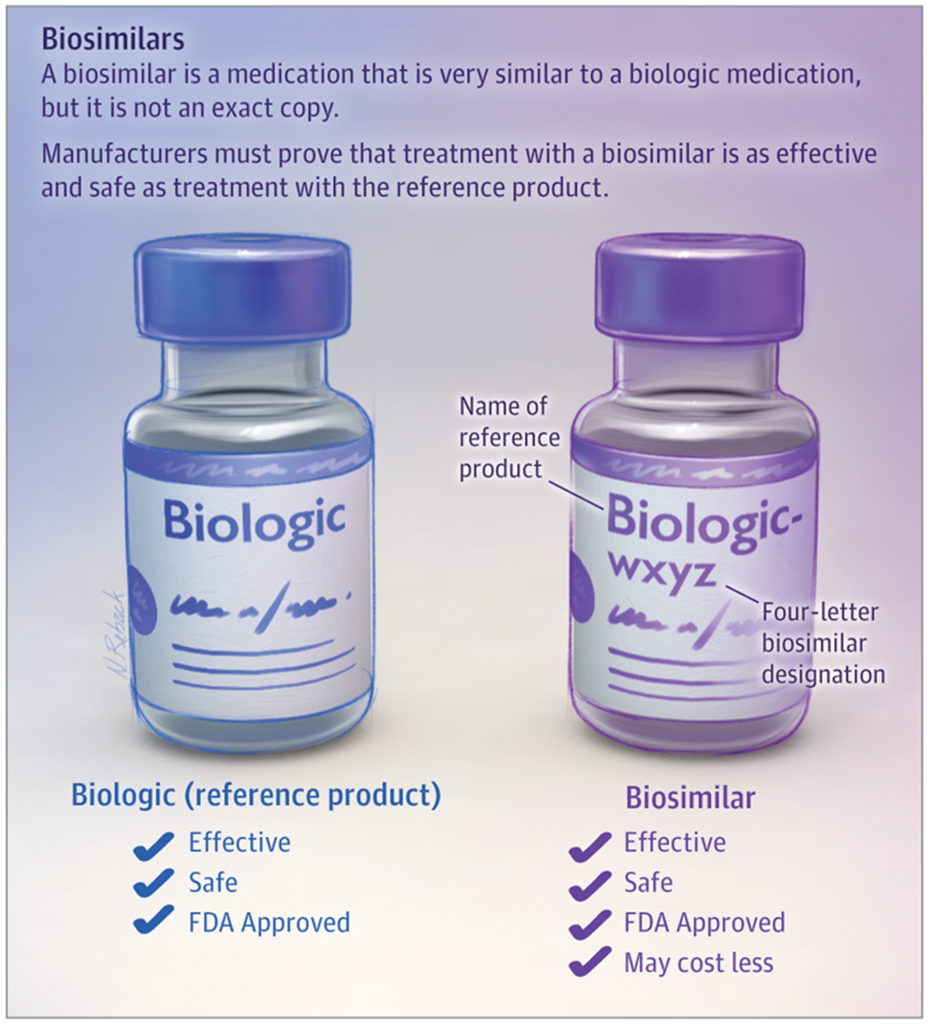
Figure 2. Biosimilars are similar to their reference biologic drugs. Source: Parsad S, Nabhan C. Biosimilars. JAMA Oncol. 2018;4(7):1023. doi:10.1001/jamaoncol.2018.0999
How do drug developers show biosimilarity?
To ensure products meet the regulatory standards of similarity, a comprehensive outlook includes a review of quality data (analytical comparability exercise), in vitro and in vivo non-clinical data, and comparative pharmacokinetic (PK), pharmacodynamic (PD), safety, immunogenicity (which is important for a biosimilar as having the same as the reference, not higher), and efficacy studies. The collective evidence should indicate no clinically meaningful safety, purity, or potency differences between the proposed biosimilar and the reference product.
In general, clinical efficacy studies aren’t required if there’s sound PK and PD biomarker data. PK similarity alone may be sufficient to support a biosimilar application.
However, drug developers commonly use PD biosimilarity when there are sensitive exposure-dependent changes in biomarkers. For example, Li et al (2018) compared the use of severe neutropenia duration and the area under effect curve of absolute neutrophil counts. They showed that absolute neutrophil count was a predictive safety biomarker.
The FDA provides more granularity on designing clinical studies to show biosimilarity. They encourage sponsors to characterize exposure and exposure/response relationships. They also provide guidance on the clinical pharmacology principles needed for a smooth review of regulatory dossiers.
In terms of selecting biomarkers for response, they offer additional specificity:
- The time of onset of change in the PD biomarker relative to dosing and its return to baseline with discontinuation of dosing
- The dynamic range of the PD biomarker over the exposure range to the biological product
- The sensitivity of the PD biomarker to differences between the proposed biosimilar product and the reference product
- The relevance of the PD biomarker to the mechanism of action of the drug (to the extent that the mechanism of action is known for the reference product)
- The analytical validity of the PD biomarker assay
Are bioequivalence standards applied to biosimilars?
Biosimilars aren’t generic small molecule drugs. As they have biological origins, ensuring pharmacodynamic similarity is vital. While regulatory guidances have suggested bioequivalence-based bounds, CI of 80-125%, such stringent thresholds are often unnecessary (read this blog to learn more about meeting bioequivalence requirements).
Health authorities have indicated that more liberal thresholds are sometimes acceptable or justifiable. Model-based arguments supporting similarity based on PD principles are generally acceptable. Refer to David Strauss’s paper for context on wider bounds.
Factoring pharmacodynamic complexity when deciding on biosimilarity
Because biosimilars are not generic drugs, it is crucial to understand the inherent biological pathways by which the biologic mediates its pharmacodynamic response. Yan and coworkers’ research on erythropoietin biosimilars exemplifies this complexity.
Erythropoietin (EPO) is a 30-kDa glycoprotein produced by fetal liver and adult kidney. EPO stimulates red blood cell production by binding to EPO receptors (EPOR) on the surface of erythroid precursor cells. Mediators that are receptor ligands can trigger a signaling pathway that either up or downregulates or changes total receptor pool size. These mediators are susceptible to pharmacodynamics-based drug disposition.
Yan et al modeled the link between the erythroid precursor cells and receptor pool for rHuEPO (recombinant human EPO) and found a proportional relationship. Their work highlights the complexity in showing that biosimilars are “highly similar” to their reference products based only on pharmacokinetics. For some biosimilars, such as epoietins, pharmacodynamic or model-based similarity should be assessed.
In conclusion, biosimilars are necessary to ensure patient access and affordability. However, demonstrating biosimilarity requires considering exposure/response relationships to ensure that the inherent biological pathways are elicited similarly to the reference product.
Learn more about how Certara experts can help support your complex biologic drug development program.
References and further reading
- European Medicines Agency, The European Commission. Biosimilars in the EU – Information guide for healthcare professionals. Prepared jointly by the European Medicines Agency and the European Commission; 2019.
- Guideline on similar biological medicinal products, CHMP/437/04 Rev 1, 23 October 2014
- Guideline on similar biological medicinal products containing biotechnology-derived proteins as active 93 substance: non-clinical and clinical issues, EMEA/CHMP/BMWP/42832/2005 Rev1, 18 December 2014
- Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1), EMA/CHMP/BWP/247713/2012, 22 May 2014
- Li, L. et al Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G‐CSF in breast cancer patients. Clin. Pharmacol. Ther. 104, 742–748 (2018).
- Silverman, E. (2005) Biologics looming price tag has payers retooling pharmacy coverage. Biotechnology Healthcare, April: 35–42.
- Strauss D et al. Pharmacodynamic Biomarkers Evidentiary Considerations for Biosimilar Development and Approval. Clin Pharmacol Ther. 2023 Jan; 113(1): 55–61.
- US Food and Drug Administration. FDA guidance: scientific considerations in demonstrating biosimilarity to a reference product. (2015).
- US Food and Drug Administration. FDA guidance: clinical pharmacology data to support a demonstration of biosimilarity to a reference product (2016).
- U.S. Food & Drug Administration. Biosimilar and Interchangeable Products.
- Yan W et al. Population Pharmacokinetic and Pharmacodynamic Model-Based Comparability Assessment of a Recombinant Human Epoetin Alfa and the Biosimilar HX575. J Clin Pharmacol. 2012; 52: 1624-1644.
