



Every FDA Advisory Committee meeting is unique, and the Cellular, Tissue, and Gene Therapies Advisory Committee meeting that took place on April 15, 2021 was not any different. The agenda included discussion of a biologics license application (BLA) for donislecel (purified allogeneic deceased donor pancreas derived Islets of Langerhans) for the treatment of brittle Type I diabetes mellitus (TlD). This, by itself, is a groundbreaking development, considering that insulin is the only treatment option that is currently available to patients with Type 1 diabetes. However, the Committee approach and outcome was a watershed moment for the future of cell and gene therapy development. There were three major points that are applicable to the development of any cell and gene therapy product:
Key Takeaway #1: Chemistry, Manufacturing and Controls (CMC)
The entirety of the morning session was devoted to the discussion of the manufacturing process and the product’s potency and purity. The scrutiny and discussion were reminiscent of the early stages of biologics development when “your process was your product” and strongly reinforced the expectation to define a product’s critical quality attributes (CQA), corelating CQA with clinical performance and establishing controls for CQAs.
Any company that is developing a cell or gene therapy must invest substantial time and effort in product development. This includes, but is not limited to, developing a testing methodology, establishing process controls, defining critical steps, and, most importantly, testing the ability of the process to perform as expected at a commercial scale.
Key Takeaway #2: Clinical Program
The time of complex clinical programs consisting of multiple studies with hundreds and sometimes even thousands of patients may be coming to the end. The primary evidence for clinical efficacy and safety that were presented to the Committee came from one Phase 1/2 and one Phase 3 single arm open-label studies. Type 1 diabetes natural history data were used as an external control. The FDA’s position was clearly presented “while clinical evidence of safety and effectiveness for licensure is often derived from prospective, randomized, controlled clinical trials, substantial evidence of effectiveness may come from a single-arm study compared to a performance goal based on well-characterized natural history of the disease, in this case metabolically unstable Type 1 diabetes “2.
The clinical development program of any cell or gene therapy product should be based on extensive knowledge of the disease and disease progression, understanding the mechanism of action, and identification of clearly defined and clinically meaningful endpoints. For diseases with a long history, like Type 1 diabetes, historical data may be used to reduce the clinical burden.
Key Takeaway # 3: Long-term Impact
There were multiple and well justified concerns regarding a) risks with long-term immunosuppression and b) duration of insulin independence. At the time of the donislecel submission, results on insulin independence for more than 1 year was reported in only 21 of 30 subjects. It is expected that there will be post-marketing commitments to monitor and further assess the long-term impact of treatment with donislecel.
A post-marketing commitment for long-term follow up and collection of real-world evidence is expected to become an important part of any cell or gene therapy approval. These commitments will allow for timely approval and access to these important, novel therapies.
The outcome of the Committee meeting was positive with a 12 to 4 vote recognizing the clinical benefit of donislecel. The Committee agreed that donislecel demonstrated a favorable risk-benefit profile for certain patients with difficult-to-control Type I diabetes.
Conclusion
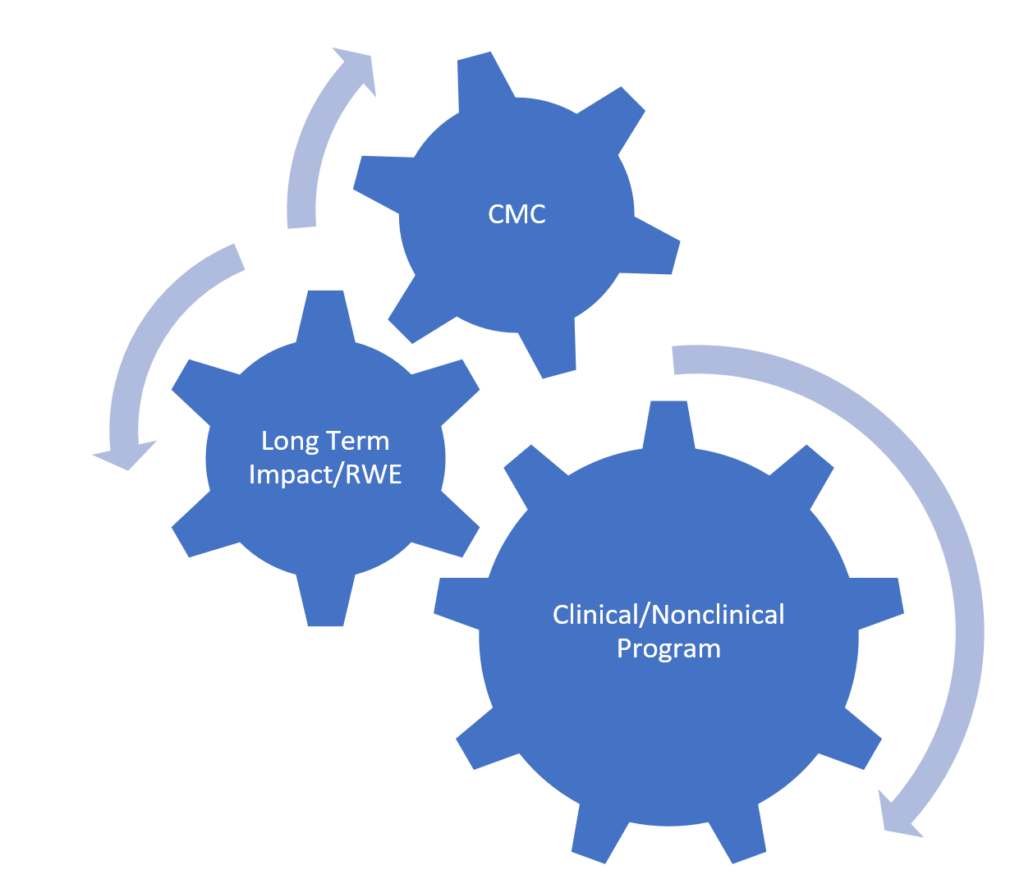
These three key takeaways should be considered at the beginning of any cell or gene therapy development program and should be organically connected, as presented in the following diagram. The best way of connecting these different aspects of development is through a well-planned and aligned regulatory and market access strategy.
Certara/Synchrogenix’s expert consultants can assist you with various aspects of your program as well as designing your regulatory and market access strategy. To learn more, please watch a webinar that we gave on this topic.
References
1. FDA Cellular, Tissue, and Gene Therapies Advisory Committee April 15, 2021 Meeting Announcement. Last accessed 23-Apr-21: https://www.fda.gov/advisory-committees/advisory-committee-calendar/cellular-tissue-and-gene-therapies-advisory-committee-april-15-2021-meeting-announcement-04152021
2. FDA Briefing Document, Cellular, Tissue, and Gene Therapies Advisory Committee Meeting April 15, 2021. BLA 125734 DONISLECEL Applicant: CellTrans, Inc. last accessed, 5-May-21: https://www.fda.gov/media/147525/download