



The regulatory landscape for drugs to treat rare diseases is being disrupted. Gleaning from several recent health authority advisory committee deliberations have provided a glimpse into the science and complexity of new drug development for rare diseases. Health authorities, drug developers, and key opinion leaders alike struggle to attribute meaningful data signals in clinical studies for rare disease indications. In this blog, I will share our 3 key considerations in ensuring maximal probability of success in rare disease development. I will refer to the Duchenne example to highlight these features.
Duchenne muscular dystrophy (DMD) is a progressive and fatal rare X-linked neuromuscular monogenetic disease with few therapeutic options. It is caused by mutations in the DMD gene that prevent the production of functional dystrophin protein. Because of the mutation, individuals with DMD produce little or no functional dystrophin in their muscle. This lack of functional dystrophin is a major cause of DMD. Multiple rare disease developers are targeting the dystrophin pathway, so significant data exists on dystrophin-related pharmacodynamics. The issue, however, is correlating increasing the levels of a target engagement biomarker such as dystrophin and meaningful clinical efficacy. To date, accelerated approval has been granted to several full-length “dystrophin” based anti-sense oligonucleotides (EXONDY 51, VYONDY 53, AMONDY 45, VILTEPSO™) as well as one “micro-dystrophin” AAV therapy (ELEVIDYS).
Ensure that your biomarker is biologically plausible.
The choice of biomarker and/or surrogate endpoint is critical to ensuring success in any rare disease program because of two reasons (read more about biomarker strategies in this white paper). One is ensuring a predictable relationship between the chosen biomarker and the clinical endpoint, and two, changes in drug response is explained by biology or biological plausibility. If you are working on a program that has a well-known surrogate endpoint, then you’re in luck. However, if you don’t have a surrogate endpoint, you’ll carry the burden of proving biological plausibility. Given that DMD is a neuromuscular disorder, drug developers of gene therapies can use the construct contained within the FDA’s Human Gene Therapy for Neurodegenerative Diseases guidance (FDA 2022) that states: ‘Use of a surrogate endpoint may be appropriate under certain circumstances such as when a [gene therapy] GT product directly targets an underlying, well-understood and well-documented monogenic change that causes a serious neurodegenerative disorder.’
Almost all treatments for DMD have included dystrophin as the biomarker of interest. This is because replacing the missing dystrophin gene in both skeletal and cardiac muscles is purported to increase the quality of life of DMD patients (Davies et al., 2019). Matching a low gene capacity of recombinant adenovirus-associated virus and the large size of the dystrophin mRNA has been challenging for gene therapy development. The recent ELEVIDYS approval showed that micro-dystrophin is also therapeutically significant and could pave the way for more gene therapy options for DMD. How such a biomarker correlates with a clinical endpoint is crucial and any studies including such novel biomarkers should define the relationship between the biomarker and the clinical endpoint. For example, the FDA guidance on DMD suggests using North Star Ambulatory Assessment (NSAA) to measure gross motor function. Other acceptable clinical measurements include timed function tests such as time to climb four stairs or time to walk or run 10 meters, in ambulatory children ages 3 years and older, the, among others.
Execute a clinical program that is best designed as a randomized controlled clinical trial.
Signal attribution to the novel treatment is always important in any new drug development program. Is the drug working as intended? Not only this is a question of biology, but it is also about how that biology was assessed. Sponsors are always quick to find easy, cheap ways to run clinical studies and may opt for open label studies without controls. It is like running an obesity clinical trial for a new drug candidate without a placebo arm. Imagine, if you had placebos in that study, you would see that there is weight loss in the placebo group due to diet and exercise requirements for all participants in the study. You may attribute the signal to the treatment if your clinical trial design wasn’t done properly. Hence, using the randomized clinical trial (RCT) to assess efficacy and safety of your new molecule, whether in common or rare diseases, is always a safe choice. RCTs are statistically superior to open label designs and are emphasized in the FDA Guidance for Industry for DMD (2018).
When you have a wide variation in pharmacology within your clinical program (e.g., the age ranges from 2 to 25 years), you might notice that you may not have the required sample size to draw inferences on the activity. One may be quick to pool data across the studies, which may be a combination of open label studies as well as randomized studies. On the other hand, one may resort to subset analyses.
Subset analyses have both advantages and disadvantages. The advantages include the possibility of developing a hypothesis. However, if one uses subset analysis to confirm a hypothesis, then there is a risk of increased probability of a statistically significant false-positive result. The more groups one investigates, the greater the probability one would find a statistically significant effect purely by chance (“p-hacking”; Head et al., 2015). To do hypothesis testing based on subgroup analyses or pooled data, pre-specify the hypothesis testing and multiplicity adjustment. The FDA has been clear that exploratory subgroup analyses are insufficient for confirmation of effectiveness.
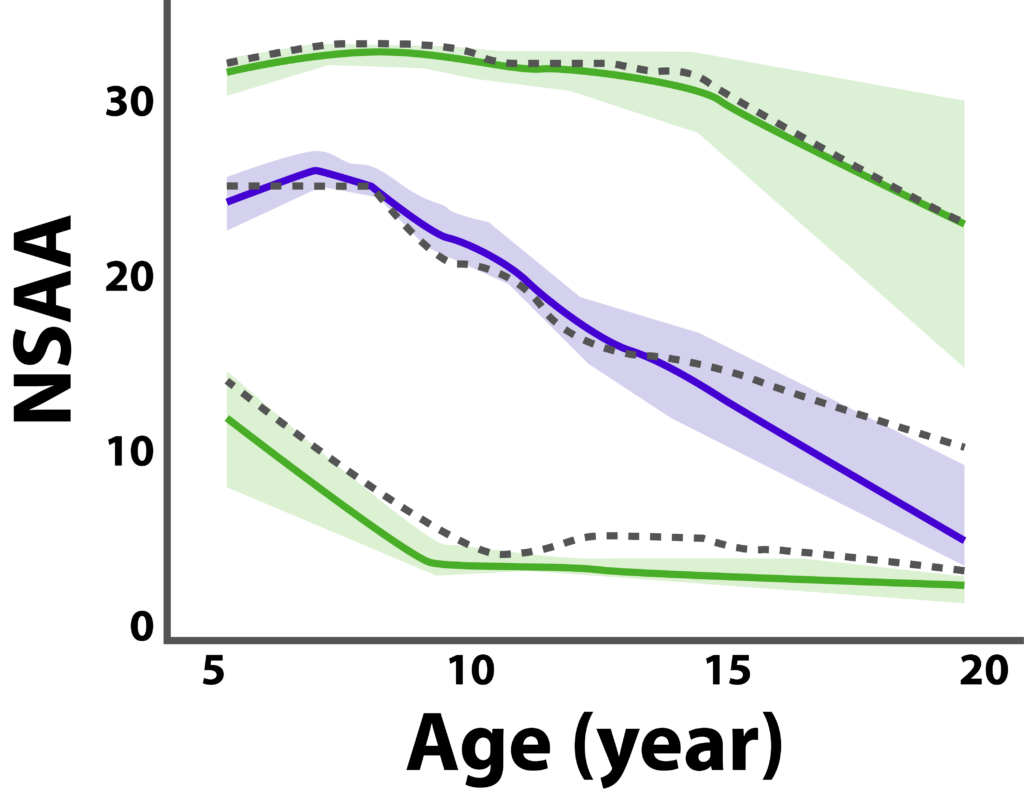
Lingineni et al have used a model-based approach to identify covariates of interest and disease progression information on DMD to optimize clinical trial designs. Figure 2 shows the profile of NSAA, a common trial endpoint in DMD studies. The visual predictive check shows the median (dashed black line) and the 5th and 95th percentiles (lower and upper dashed lines, respectively) of the observed data, whereas the blue shaded areas indicate the 90% CIs of the model prediction of the median, and the green shaded areas show 90% CIs of the model prediction for the 5th and 95th percentiles. The solid lines represent the model prediction.
Utilize the value of external controls in your clinical program.
Whilst there is emerging regulatory interest and acceptance of external controls in clinical development programs, their use is often variable and inconsistent with best practices. Consequently, use of such controls is often viewed as supportive evidence. Rare disease developers using integrated analyses with external controls are using such approaches in study level and integrated analyses. Some use the propensity score weighting method to select external control subjects with greater similarity to the subjects enrolled in their clinical studies. These external control data are typically obtained from available libraries of natural history cohorts.
For DMD, sponsors typically obtain these data from the Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS) or the Finding the Optimum Regimen for Duchenne Muscular Dystrophy (FOR-DMD) clinical study. There are many ongoing NH trials (e.g., NCT03882827). The challenge, however, is finding consistency between the applied inclusion criteria with the demographics and characteristics of subjects enrolled in the sponsor drug effect trials.
External control comparisons contain several limitations. They include the heterogenous nature of the disease course and potential for non-comparability, the role of bias in characterizing the intended treatment effect, and the true similarity of the external population with the trial population. The focus here is on unobserved or unquantifiable baseline characteristics. Something like the propensity score weighting method is also subject to unverifiable assumptions regarding confounding factors.
For more information on how model-informed drug development can meaningfully move the needle on rare diseases therapeutics, please refer to the author’s editorial in the Journal of PK and PD.
References
- Davies KE et al. Micro-dystrophin Genes Bring Hope of an Effective Therapy for Duchenne Muscular Dystrophy. Molecular Therapeutics. 2019 Mar 6; 27(3): 486–488.
- FDA Guidance, Duchenne Muscular Dystrophy and Related Dystrophinopathies: Developing Drugs for Treatment Guidance for Industry, February 2018.
- Head ML et al. The Extent and Consequences of P-Hacking in Science. PLoS Biology. 2015 Mar; 13(3): e1002106
- Lingineni K, Aggarwal V, Morales JF, et al. Development of a model-based clinical trial simulation platform to optimize the design of clinical trials for Duchenne muscular dystrophy. CPT Pharmacometrics Syst Pharmacol. 2022;11(3):318-332.
- Wang H et al. Clinical Trials with External Control: Beyond Propensity Score Matching. Statistics in Biosciences. 2022, 14, 304–317.
