



New therapeutics discovery and development for ocular diseases have been traditionally associated with a low probability of technical and regulatory success. The significant unmet medical needs for ocular diseases include but aren’t limited to glaucoma, age-related macular degeneration, and retinopathy. Recently, there has also been considerable focus on thyroid eye disease (TED). This research gained impetus through the 2020 approval of Horizon Pharma’s teprotumumab-trbw (Tepezza®) for treating TED (FDA press release, 2020). Interestingly, teprotumumab was initially evaluated in multiple oncology indications prior to its development and approval in TED.
Figure 1. Anatomy of the eye and surrounding tissue.
TED is a serious, rare, and debilitating autoimmune disease, which, if left untreated in severe illness, can result in vision impairment (National Organization for Rare Disorders; NORD). Also known as thyroid-associated ophthalmopathy, Graves’ eye disease, or Graves’ orbitopathy (Figure 2). TED causes progressive inflammation and damage to extraocular muscle, connective, and fatty tissues. This damage results in proptosis (eye bulging) can have a significant impact on emotional well-being and quality of life. It is more common in women than men with a median age at diagnosis of 43 years. Consequently, risk factors for TED include female gender, middle age, and smoking. While the etiology of TED continues to be a well-researched subject, patients appear to overexpress the insulin-like growth factor-1 receptor (IGF-1R), which plays a principal role in the development of the disorder (Douglas et al., 2008). The disease meets the criteria of rare diseases, and NORD indicates a prevalence rate of 16 per 100,000 women and 2.9 per 100,000 men. Therapies for TED have ranged from corticosteroids to orbital radiation.
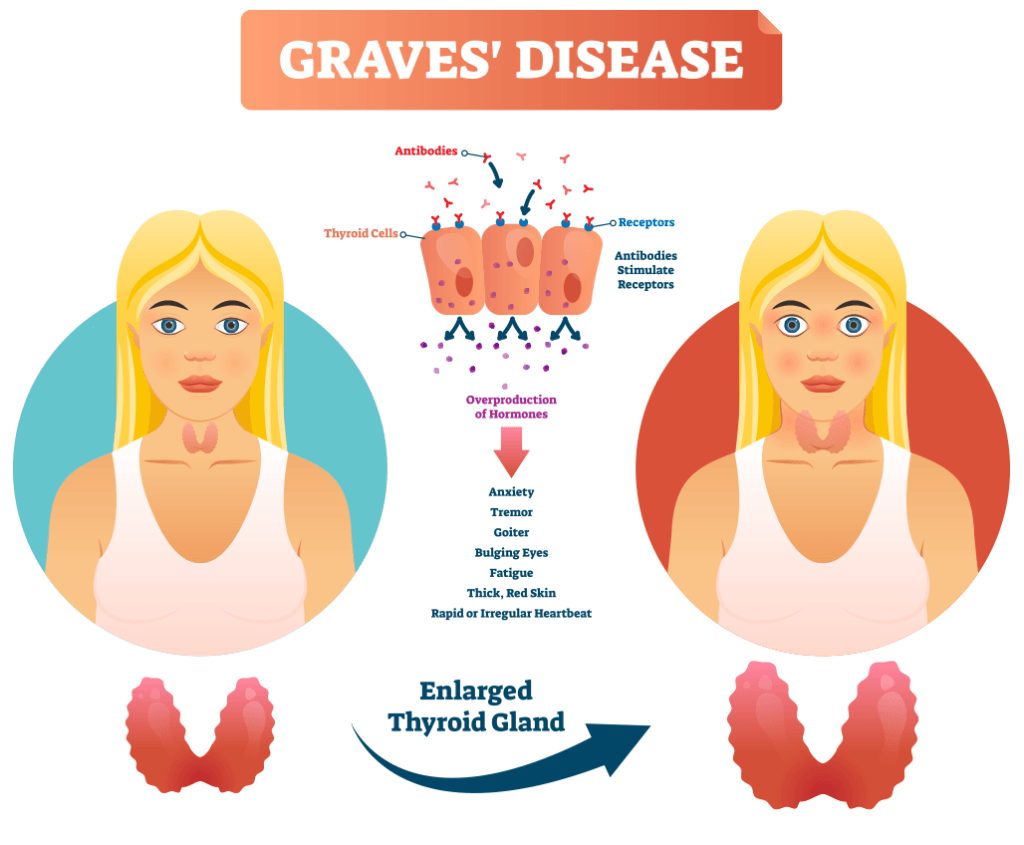
Figure 2. Graves’ Disease pathology
Tepezza is a biologic, a fully human monoclonal antibody (mAb), which inhibits IGF-1R. This approval deviates from the central philosophy in ocular therapeutics development, which is predicated on effect site compartment access for small molecules. Tepezza is a systemically-acting biologic. Let’s reflect on this approval and some of the key considerations that affect most ocular drug development programs. The US FDA conferred Tepezza with orphan drug status while granting a plethora of regulatory pathways that included priority review, fast track, and breakthrough therapy designations.
Here are three key considerations.
Access to ocular tissues for optimal effect
Whereas teprotumumab development reflected the development of a systemic acting biologic, there are many ocular therapies in development that involve small molecules. In such instances, there are many tools in the armamentarium to think about. Principally, they involve delivering the investigational agent to the eye topically, systemically, or injecting into the ocular or periocular regions. While topical administration represents the simplest approach, it is perhaps the least effective of the three as penetration into the effect site may be limiting. Systemic small molecule agents often need higher active concentrations to reach the site of action and could have limiting safety effects. The last mode of delivery is invasive but effectively lands the molecule in the region of effect.
Designing an effective molecule with an intended mode of delivery that is matched to the intended target compartment is a necessary first step in ocular drug discovery programs.
Importance of discerning effect in a small clinical study
Perhaps the most critical challenge in rare disease development drug is the inherent small patient population. In the clinical development of teprotumumab, efficacy was determined by the ability of the drug to induce a clinically meaningful reduction in proptosis (FDA SBA, 2021).
A review of the summary basis of approval of Tepezza revealed that 82% of patients treated with 8 doses of teprotumumab exhibited at least a 2-millimeter reduction in proptosis compared to only 16% of patients treated with placebo. A 2-millimeter reduction was considered to be a clinically significant threshold as it was believed to be necessary to reduce the incidence of diplopia (double vision) and improve the lid coverage over the cornea. The clinical program included 120 patients, which is typical of a rare disease program. Interestingly, there appeared to be no meaningful trend in exposure-PRR (proptosis responder rate) relationship in the efficacy cohort. The primary adverse events of teprotumumab in TED patients were hyperglycemia and muscle spasm, which again revealed no notable exposure-safety relationships. Because of the small sample size and a single dosing regimen, lack of exposure-response relationships for efficacy and safety usually does not serve as a hypothesis generating exercise.
Because of the small sample size and the design of the study, there was no way to understand the durability of response or frequency of relapse. A long-term follow up assessment is not available within the approval dossier, although the sponsor notes there is an extension study in progress. Modeling the disease progression will likely reveal mechanistic insights into the response durability.
Another challenge in the management of small clinical trials is the selection of the endpoints (EMA, 2006). While the selection depends on the acceptance of a regulatory pathway of interest, there are a few aspects to think about. The endpoint that reflects a certain clinical benefit (e.g., overall survival) is one that meets the regulatory definition of endpoints without much question. However, an earlier temporal effect may also be a choice, subject to it meeting the statutory definitions and appropriate validations. If pursuing time to disease progression as an endpoint, measuring disease severity is necessary with appropriate follow-up studies. Renal failure as a clinically relevant endpoint may be justified in Fabry disease as it affects patient survival (EMA, 2006).
Dose selection
Another key consideration in the development of drugs for rare diseases is dose selection. Because a significant amount of information is usually generated through drug repurposing, there is usually some information available for the investigational agent if it was assessed in non-rare indications. For example, intensive pharmacokinetic (PK) data were generated from 36 patients with advanced solid tumors, non-Hodgkin’s lymphoma, or Hodgkin’s lymphoma in the Tepezza development program, which the sponsor pooled with the sparse teprotumumab PK data from TED patients.
In the case of teprotumumab dosing, the rationale for regimen selection was made based on the degree of saturation of target mediated clearance in TED patients, i.e., a target of >90% was selected (Xin et al., 2021). Interestingly, this determination was based on a dose ranging study done from 1 to 16 mg/kg in oncology patients. The summary basis of approval revealed that neither IGF-1 concentrations nor estimated IGF-1R saturation thresholds were available in TED patients. There were also limitations in the IGF-1R/IGF-1 levels vs. efficacy response analysis that could lend some validation of efficacy in TED patients. Notwithstanding these caveats, the dosing regimen taken forward in the study in TED patients revealed clinically meaningful improvements. Later, a publication by the sponsor revealed how model-informed drug development (MIDD) principles were applied in dose selection (Figure 3).
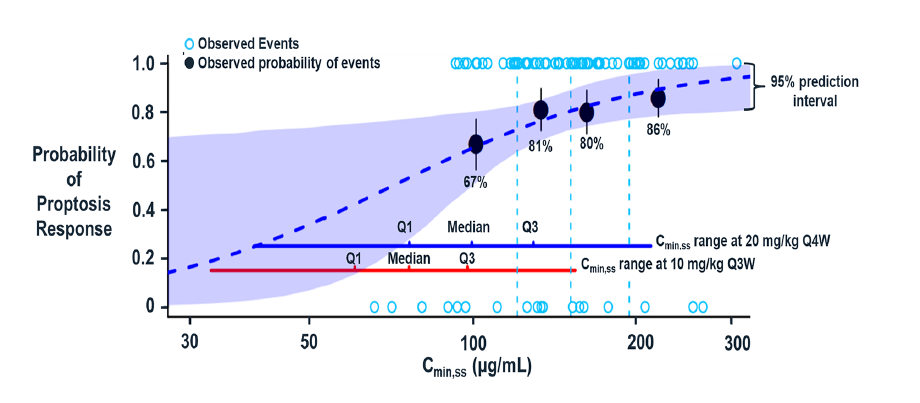
Figure 3: Xin et al show the probability of proptosis response at week 24 vs steady-state trough concentration (Cmin,ss) [Adapted from Xin et al., 2021]
The role of biomarkers and surrogate endpoints in the natural progression of disease is a critical determinant of the clinical development process for rare diseases. Choosing a biomarker that is expected to be predictive of the clinical benefit is a key consideration.
In summary, the development of ocular drugs for both common and rare diseases have important implications for sponsors. Ensuring effective understanding of disease progression, biomarkers, and choice of endpoints, as well as ensuring that precision around the effect meets regulatory muster are critical determinants of technical and regulatory success.
Certara, with its tech-enabled services and expertise in MIDD and rare diseases, is well positioned to advise on rare and common disease development programs. Contact us for your needs. For a glimpse into the role of model-based meta-analysis in the development of ocular therapies, please read this publication:
References
- FDA press release (2020). FDA approves first treatment for thyroid eye disease. URL: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-thyroid-eye-disease (Last accessed: November 29, 2021).
- FDA Summary basis of approval, Drug Approval Package: TEPEZZA. URL: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761143Orig1s000TOC.cfm (Last accessed: November 29, 2021).
- NORD. Thyroid Eye Disease – NORD (National Organization for Rare Disorders). URL: https://rarediseases.org/rare-diseases/thyroid-eye-disease/ (Last accessed: November 29, 2021).
- Douglas RS, Naik V, Hwang CJ, et al. B cells from patients with Graves’ disease aberrantly express the IGF-1 receptor: implications for disease pathogenesis. J Immunol 2008; 181: 5768-74.
- EMA 2006. Guideline on clinical studies in small populations. URL: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761143Orig1s000TOC.cfm (Last accessed: November 29, 2021).
- Xin Y et al. Pharmacokinetics and Exposure‑Response Relationship of Teprotumumab, an Insulin‑Like Growth Factor‑1 Receptor‑Blocking Antibody, in Thyroid Eye Disease. Clinical Pharmacokinetics (2021) 60:1029–1040.
