



A symbiosis between a modeler and a trialist is often required to develop a model-informed drug development strategy. Modelers can design an experiment so the results can be interpreted in a more integrative fashion. The modeler often views the totality of the data from multiple lines of investigation while the trialist is likely focused on the immediate trial.
The way that modeler and trialist roles are set up in any R&D organization often determines the seamlessness in executing such a strategy. In some organizations, these roles are combined into a single department.
But frequently, multiple departments handle these roles. For example, the trialist could be from a clinical pharmacology, experimental medicine, or clinical development function. Likewise, the mathematical modeler could be sourced from a quantitative science, data sciences, biostatistics, or pharmacometrics (PMX) function.
If you are a part of the latter structure, then this blog will likely interest you as we highlight three key areas where integrating modeling and trial science adds value.
#1 Understanding drug response and mechanism of drug action
This is quite an important area where the two disciplines need to work together. Identifying what constitutes a drug response, how that is measured, and ensuring that the response being modeled (i.e., in silico drug testing) is a direct measure of the pharmacology is key.
In early clinical trials, investigators may observe many responses. They may range from biomarkers, mechanistic effect (e.g., enzyme inhibition), potential or accepted surrogate measures (e.g., blood pressure, lipoproteins, etc.), to clinically remote measures (e.g., receptor occupancy). Ensuring that trials collect the right information and understanding their value for overall biology is critical.
Depending on the effect assessed, it will be helpful to consider whether the effect is derived from a single dose, at steady state, or at the end of the dosing interval. This will ensure that you choose the right modeling method. Time dependencies may need to be assessed if the effects are delayed compared to exposure.
Modeling can be used to interrogate the data to ensure that assumptions regarding the time course of the effect are valid. A variety of other time-related effects on drug action such as induction, tolerance, and chronopharmacology could influence the way PMX analyses are set up. This is where clinical pharmacology and modelers must collaborate!
#2 Determining the optimal study design for the question at hand
Often, sponsors conduct clinical studies using trial designs that are more “fit for purpose” and address the immediate issue at hand. The choice of study design could affect the way PMX analyses are set up. Before each study design discussion, the modeler and the trialist should collaborate to determine the optimal study design with the best operating characteristics that would optimally inform planned analyses. The FDA exposure/response guideline reflects nicely on such choices.
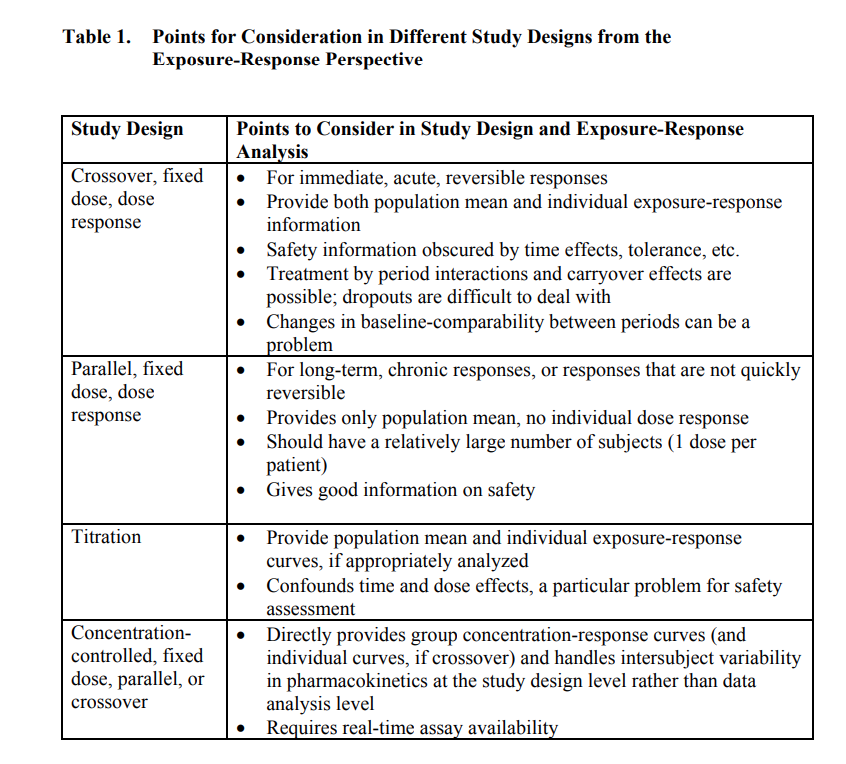
Another area for trialist-modeler collaboration is selecting the dose and dosing regimen. Traditionally, clinicians selected doses by gestalt. Often, this meant selecting a maximum tolerated or feasible dose, such that a single effect is elucidated.
This is not a viable strategy from a “learn and confirm” perspective. Often, we need to understand the dynamic range of effects as a function of a range of dose levels. A modeler may suggest a more informative dose paradigm to identify the minimum effective dose or a dose that can yield desirable clinical outcomes or a range to interpolate a dose for further study. Discussing the method of dose selection is often valuable compared to deterministic dose selection.
In this context, there are a couple of other aspects worth noting. One relates to the analytes measured and the other to the pharmacokinetic (PK) endpoints. Regarding analytes, ensure both parent and metabolites are measured in the clinical studies. Metabolites aren’t optional!
Important considerations for metabolite assessments include whether they are active, toxic (in pre-clinical studies, in which case monitoring is essential), have downstream pharmacology for delayed effects, and whether they may explain altered pharmacodynamics. Increase your efficiency by including metabolites in the PMX analyses.
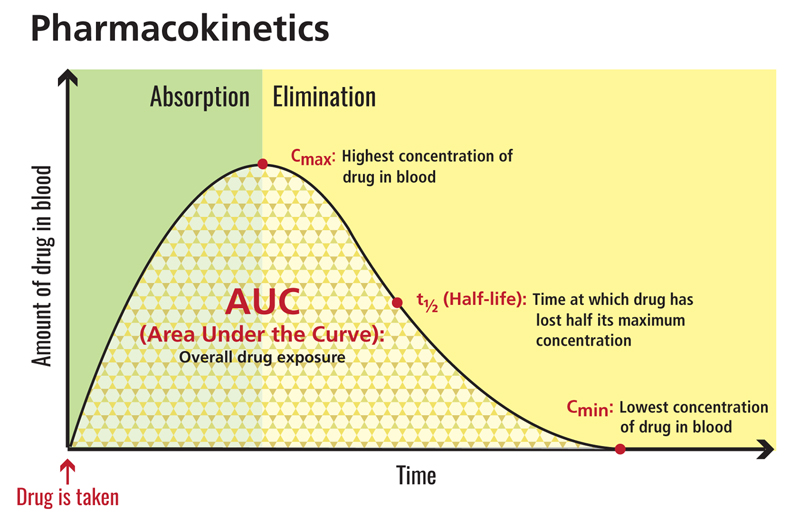
Another common area of consideration in any analysis includes the PK parameter of interest. Often, AUC, Cmax, and Cmin are pursued. When selecting an endpoint, identify what parameter best tracks efficacy and safety. Remember some safety endpoints track better with Cmax (e.g., vital signs) and some better with AUC (e.g., hepatic enzymes).
#3 Assessing the clinical meaningfulness of a change
Deciding on a threshold for a clinically significant effect can be confusing! All new small molecule drug or biologic development programs must understand the threshold for clinical significance of the effect. This is key because of posology considerations but also package insert considerations.
One normally starts with the easiest of available thresholds, the “bioequivalence bounds”. These are very stringent bounds (acceptance intervals of 0.8-1.25) used in formulation specifications studies where such stringency in product performance is expected.
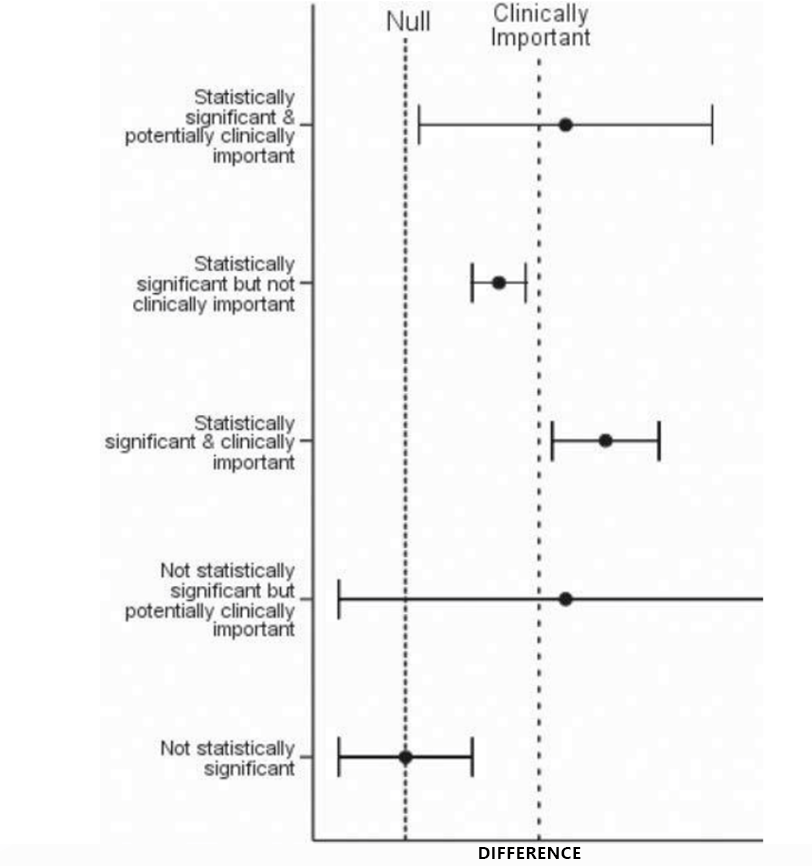
However, such narrow bounds are typically unnecessary for a safe and well-tolerated drug. Health authorities are always open to the use of wider bounds if justified. Ensuring the understanding of the dose margins (e.g., a clinical dose of 10 mg vs the highest dose studied of 100 mg) and whether there are dose-limiting safety aspects or saturable effects due to the solubility of the drug substance would be essential. Modelers can help determine this threshold by several aggregate and population-based methods, including, but not limited to model-based meta-analysis (MBMA).
Another area for collaboration is determining the worst-case effect for a given dose or dosing regimen. For example, a sponsor developing a drug with a blood pressure adverse effect might ask, “What is the risk that the drug will result in x% high blood pressure?” or “What is the worst QTc prolongation effect that could occur with this drug?”
These questions deserve the input of trialists and modelers. The underlying mechanism of effect and not just the effect itself (i.e., what you see) is important to understand.
This is embodied in the extreme value theory that has been in use since the early 1900s. A good articulation of the use of extreme value theory in pharmacometrics is eloquently described in the work by Bonate, 2020. Therefore, any modeling analysis should characterize the variability in response and not just mean trends.
In summary, executing a model-informed drug development strategy requires a close connection and, ideally, a seamless integration of the trialist and modeler functions. They can collectively maximize the value harnessed from model-informed drug development approaches in the selection of the right dose, the right study population, the right endpoints, and the right registration strategy.
To get our expert insights on creating a credible biomarker strategy for accelerated drug development and approval, check out this white paper.
This blog was originally published on January 6, 2023. It was updated on November 8, 2024.
References
- Osuolale, Kazeem. (2020). Confidence Interval as a Better Alternative to P-Value for Clinical Significance. 47-51. 10.9790/5728-1603044751.
- US FDA Exposure-Response Guidance. (Last accessed: September 16, 2022)

